|
 |
Para ver los documentos PDF marcados con hipervínculo se requiere Adobe Acrobat Reader.

Se puede descargar localmente el archivo PDF haciendo uso del botón derecho del ratón.
|
 |
Modulation of taxane binding to tubulin curved and straight conformations by systematic 3'N modification provides for improved microtubule binding, persistent cytotoxicity and in vivo potency.
Ma Y1, Josa-Prado F2, Essif JN3, Liu S4, Li S1, Lucena-Agell D2, Chan PY3, Goossens K2, Hortigüela R2, Matesanz R2, Wang Y4, Gago F5, Wang H, Risinger A3, Díaz JF6, Fang WS7
Author information:
1 State Key Laboratory of Bioactive Substance and Function of Natural Medicines & Ministry of Health Key Laboratory of Biosynthesis of Natural Products, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, 2A Nan Wei Road, Beijing, 100050, China. 2 Centro de Investigaciones Biológicas Margarita Salas, Consejo Superior de Investigaciones Científicas, Ramiro de Maeztu 9, Madrid, 28040, Spain. 3Department of Pharmacology, University of Texas Health Science Center at San
Antonio, San Antonio, TX, 78229, United States. 4 Key Laboratory of Molecular Pharmacology and Drug Evaluation (Yantai University), Ministry of Education, Collaborative Innovation Center of Advanced Drug Delivery System and Biotech Drugs in Universities of Shandong, Yantai
University, Yantai, 264005, China. 5 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al Instituto de Química Médica del CSIC, Universidad de Alcalá, E-28805, Alcalá de Henares, Madrid, Spain. 6 Centro de Investigaciones Biológicas Margarita Salas, Consejo Superior de Investigaciones Científicas, Ramiro de Maeztu 9, Madrid, 28040, Spain. 7 State Key Laboratory of Bioactive Substance and Function of Natural Medicines & Ministry of Health Key Laboratory of Biosynthesis of Natural Products, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College, 2A Nan Wei Road, Beijing, 100050, China.
The taxane class of microtubule stabilizers are some of the most effective and widely used chemotherapeutics. The anticancer activity of taxanes arises from their ability to induce tubulin assembly by selectively recognizing the curved (c-) conformation in unassembled tubulin as compared to the straight (s-) conformation in assembled tubulin. We first designed and synthesized a series of 3'N-modified taxanes bearing covalent groups. Instead of discovering covalent taxanes, we found a series of non-covalent taxanes 2, in which the 3'N side chain was found to be essential for cytotoxicity due to its role in locking tubulin in the s-conformation. A representative compound bearing an acrylamide moiety (2h) exhibited increased binding affinity to the unassembled tubulin c-conformation and less cytotoxicity than paclitaxel. Further exploration of chemical space around 2h afforded a new series 3, in which derivatives such as 3l bind more tightly to both the s- and c-conformations of tubulin compared to paclitaxel, leading to more efficient promotion of tubulin polymerization and a greater persistence of in vitro efficacy against breast cancer cells after drug washout. Although 3l also had improved in vivo potency as compared to paclitaxel, it was also associated with increased systemic toxicity that required localized, intratumoral injection to observe potent and prolonged antitumor efficacy.
 PMID: 37490800
PMID: 37490800  10.1016/j.ejmech.2023.115668
10.1016/j.ejmech.2023.115668
Molecular basis of the final step of cell division in Streptococcus pneumoniae
Martínez-Caballero S1, Freton C2, Molina R1, Bartual SG1, Gueguen-Chaignon V3, Mercy C2, Gago F4, Mahasenan KV5, Muñoz IG6, Lee M5, Hesek D5, Mobashery S5, Hermoso JA7, Grangeasse C8.
Affiliations:
1 Department of Crystallography and Structural Biology, Instituto de Química-Física "Rocasolano," Consejo Superior de Investigaciones Científicas, Madrid, Spain.
2 Molecular Microbiology and Structural Biochemistry, UMR 5086, Université de Lyon, CNRS, Lyon, France.
3 SFR Biosciences, Université de Lyon, CNRS UAR3444, ENS de Lyon, INSERM US8, Lyon, France.
4 Department of Biomedical Sciences & Instituto de Química Médica-CSIC Associated Unit, School of Medicine and Health Sciences, University of Alcalá, 28805 Alcalá de Henares, Spain.
5 Department of Chemistry and Biochemistry, University of Notre Dame, Notre Dame, IN 46556, USA.
6 Structural Biology Program, Spanish National Cancer Research Center (CNIO), 28029 Madrid, Spain.
7 Department of Crystallography and Structural Biology, Instituto de Química-Física "Rocasolano," Consejo Superior de Investigaciones Científicas, Madrid, Spain.
8 Molecular Microbiology and Structural Biochemistry, UMR 5086, Université de Lyon, CNRS, Lyon, France
Bacterial cell-wall hydrolases must be tightly regulated during bacterial cell division to prevent aberrant cell lysis and to allow final separation of viable daughter cells. In a multidisciplinary work, we disclose the molecular dialogue between the cell-wall hydrolase LytB, wall teichoic acids, and the eukaryotic-like protein kinase StkP in Streptococcus pneumoniae. After characterizing the peptidoglycan recognition mode by the catalytic domain of LytB, we further demonstrate that LytB possesses a modular organization allowing the specific binding to wall teichoic acids and to the protein kinase StkP. Structural and cellular studies notably reveal that the temporal and spatial localization of LytB is governed by the interaction between specific modules of LytB and the final PASTA domain of StkP. Our data collectively provide a comprehensive understanding of how LytB performs final separation of daughter cells and highlights the regulatory role of eukaryotic-like kinases on lytic machineries in the last step of cell division in streptococci.
PMID: 37418323 10.1016/j.celrep.2023.112756
Structural insight into pharmacological stabilization of microtubules by taxanes
Prota AE1, Lucena-Agell D2, Ma Y3, Estévez-Gallego J2, Li S3, Bargsten K4, Josa-Prado F2, Altmann K-H5, Gaillard N4, Kamimura S6, Mühlethaler T1, Gago F7, Oliva MÁ2, Steinmetz MO4, Fang WS3, Díaz JF.2
Affiliations:
1 Laboratory of Biomolecular Research, Paul Scherrer Institute, Villigen PSI, Switzerland.
2 Structural and Chemical Biology, CSIC-Centro de Investigaciones Biologicas, Madrid, Spain.
3 Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China.
4 Laboratory of Biomolecular Research, Paul Scherrer Institute, Villigen, Switzerland.
5 Department of Chemistry and Applied Biosciences, ETH Zurich, Zürich, Switzerland.
6 Department of Biological Sciences, Chuo University, Tokyo, Japan.
7 Department of Biomedical Sciences, University of Alcalá, Madrid, Spain.
Paclitaxel (Taxol®) is a taxane and a first-line chemotherapeutic drug that stabilizes microtubules. While the interaction of paclitaxel with microtubules is well described, the current lack of high-resolution structural information on a tubulin-taxane complex precludes a comprehensive description of the binding determinants that affect the drug's mechanism of action. Here, we solved the crystal structure of the core baccatin III moiety of paclitaxel lacking the C13 side chain in complex with tubulin at 1.9 Å resolution. Based on this information, we engineered two tailor-made taxanes with modified C13 side chains, solved their crystal structures in complex with tubulin, and analyzed their effects along with those of paclitaxel, docetaxel, and baccatin III on the microtubule lattice by X-ray fiber diffraction. We then compared high-resolution structures of ligand-bound tubulin and microtubule complexes with apo forms and used molecular dynamics simulations to understand the consequences of taxane binding to tubulin as well as to simplified protofilament and microtubule-lattice models. Our combined approach enlightens three mechanistic questions. Firstly, taxanes bind better to microtubules as compared to unassembled tubulin due to a dual structural mechanism: Tubulin assembly imposes a preorganization of the M-loop of β-tubulin, which otherwise occludes ligand access to the taxane site, while the bulky C13 side chains preferentially recognize the microtubule-assembled over the unassembled conformational state of tubulin. Second, the occupancy of the taxane site by a ligand has no influence on the straightness of tubulin protofilaments. Finally, binding of the taxane core to the taxane site displaces the S9-S10 loop of β-tubulin resulting in microtubule expansion. Our results provide detailed insight into the microtubule-stabilization mechanism of taxanes.
PMID: 36876916 10.7554/eLife.84791
 |
Structures reported in this article: 8BDE (T2R-TTL-BacIII), 8BDF (T2R-TTL-2a), and 8BDF (T2R-TTL-2b) |
[Additional files]
Insertion of an amphipathic linker in a tetrapodal tryptophan derivative leads to a novel and highly potent entry inhibitor of enterovirus A71 clinical isolates
Martí-Marí O1, Abdelnabi R2, Schols D2, Neyts J2, Camarasa MJ1, Gago F3, San-Félix A1
Author information:
1Instituto de Química Médica (IQM, CSIC), E-28006 Madrid, Spain. 2Laboratory of Virology and Chemotherapy, Rega Institute for Medical Research, Department of Microbiology and Immunology, University of Leuven, B-3000 Leuven, Belgium. 3Departamento de Ciencias Biomédicas y Unidad Asociada IQM-UAH, Universidad de Alcalá, E-28805 Alcalá de Henares, Spain
AL-471, the leading exponent of a class of potent HIV and enterovirus A71 (EV-A71) entry inhibitors discovered in our research group, contains four l-tryptophan (Trp) units bearing an aromatic isophthalic acid directly attached to the C2 position of each indole ring. Starting from AL-471, we (i) replaced l-Trp with d-Trp, (ii) inserted a flexible linker between C2 and the isophthalic acid, and (iii) substituted a nonaromatic carboxylic acid for the terminal isophthalic acid. Truncated analogues lacking the Trp motif were also synthesized. Our findings indicate that the antiviral activity seems to be largely independent of the stereochemistry (l- or d-) of the Trp fragment and also that both the Trp unit and the distal isophthalic moiety are essential for antiviral activity. The most potent derivative, 23 (AL-534), with the C2 shortest alkyl urea linkage (three methylenes), showed subnanomolar potency against different EV-71 clinical isolates. This finding was only observed before with the early dendrimer prototype AL-385 (12 l-Trp units) but remained unprecedented for the reduced-size prototype AL-471. Molecular modeling showed the feasibility of high-affinity binding of the novel l-Trp-decorated branches of 23 (AL-534) to an alternative site on the VP1 protein that harbors significant sequence variation among EV-71 strains.
PMID: 36834952 10.3390/ijms24043539
Computational approaches to enzyme inhibition by marine natural products in the search for new drugs.
Author information: Department of Biomedical Sciences & IQM-CSIC Associate Unit, School of Medicine and Health Sciences, University of Alcalá, E-28805 Madrid, Alcalá de Henares, Spain.
The exploration of biologically relevant chemical space for the discovery of small bioactive molecules present in marine organisms has led not only to important advances in certain therapeutic areas, but also to a better understanding of many life processes. The still largely untapped reservoir of countless metabolites that play biological roles in marine invertebrates and microorganisms opens new avenues and poses new challenges for research. Computational technologies provide the means to (i) organize chemical and biological information in easily searchable and hyperlinked databases and knowledgebases; (ii) carry out cheminformatic analyses on natural products; (iii) mine microbial genomes for known and cryptic biosynthetic pathways; (iv) explore global networks that connect active compounds to their targets (often including enzymes); (v) solve structures of ligands, targets, and their respective complexes using X-ray crystallography and NMR techniques, thus enabling virtual screening and structure-based drug design; and (vi) build molecular models to simulate ligand binding and understand mechanisms of action in atomic detail. Marine natural products are viewed today not only as potential drugs, but also as an invaluable source of chemical inspiration for the development of novel chemotypes to be used in chemical biology and medicinal chemistry research.
PMID: 36827141 10.3390/md21020100
Alternative approaches to understand microtubule cap morphology and function
Oliva MÁ, Gago F, Kamimura S, Díaz JF.
Microtubules (MTs) are essential cellular machines built from concatenated αβ-tubulin heterodimers. They are responsible for two central and opposite functions from the dynamic point of view: scaffolding (static filaments) and force generation (dynamic MTs). These roles engage multiple physiological processes, including cell shape, polarization, division and movement, and intracellular long-distance transport. At the most basic level, the MT regulation is chemical because GTP binding and hydrolysis have the ability to promote assembly and disassembly in the absence of any other constraint. Due to the stochastic GTP hydrolysis, a chemical gradient from GTP-bound to GDP-bound tubulin is created at the MT growing end (GTP cap), which is translated into a cascade of structural regulatory changes known as MT maturation. This is an area of intense research, and several models have been proposed based on information mostly gathered from macromolecular crystallography and cryo-electron microscopy studies. However, these classical structural biology methods lack temporal resolution and can be complemented, as shown in this mini-review, by other approaches such as time-resolved fiber diffraction and computational modeling. Together with studies on structurally similar tubulins from the prokaryotic world, these inputs can provide novel insights on MT assembly, dynamics, and the GTP cap.
PMID: 36743020 10.1021/acsomega.6b00317
Chemical modulation of microtubule structure through the laulimalide/peloruside site
Estévez-Gallego J1, Álvarez-Bernad B1, Pera B1, Wullschleger C2, Raes O3, Menche D4, Martínez JC5, Lucena-Agell D1, Prota AE6, Bonato F1, Bargsten K6, Cornelus J3, Giménez-Abián JF1, Northcote P7, Steinmetz MO8, Kamimura S9, Altmann KH2, Paterson I4, Gago F10, Van der Eycken J3, Díaz JF1, Oliva MÁ11.
Author information:
1Centro de Investigaciones Biológicas Margarita Salas - Consejo Superior de Investigaciones Científicas, Madrid 28040, Spain. 2Department of Chemistry and Applied Biosciences, Institute of Pharmaceutical Sciences - ETH Zurich, Zürich 8093, Switzerland. 3Department of Organic and Macromolecular Chemistry, Ghent University, Gent
9000, Belgium. 4Yusuf Hamied Department of Chemistry, University of Cambridge, Cambridge CB2 1EW, UK. 5ALBA Synchrotron, CELLS, Cerdanyola del Vallés 08290, Spain. 6Laboratory of Biomolecular Research, Division of Biology and Chemistry, Paul Scherrer Institut, Villigen 5232, Switzerland. 7Ferrier Research Institute, University of Wellington, Lower Hutt 5010, New Zealand. 8Laboratory of Biomolecular Research, Division of Biology and Chemistry, Paul Scherrer Institut, Villigen 5232, Switzerland; University of Basel, Biozentrum, Basel 4056, Switzerland. 9Department of Biological Sciences, Faculty of Science and Engineering, Chuo University, Tokyo 192-0393, Japan. 10Department of Biomedical Sciences and Associated Unit IQM-UAH, Universidad de Alcalá, Alcalá de Henares 28805, Spain. 11Centro de Investigaciones Biológicas Margarita Salas - Consejo Superior de Investigaciones Científicas, Madrid 28040, Spain.
Taxanes are microtubule-stabilizing agents used in the treatment of many solid tumors, but they often involve side effects affecting the peripheral nervous system. It has been proposed that this could be related to structural modifications on the filament upon drug binding. Alternatively, laulimalide and peloruside bind to a different site also inducing stabilization, but they have not been exploited in clinics. Here, we use a combination of the parental natural compounds and derived analogs to unravel the stabilization mechanism through this site. These drugs settle lateral interactions without engaging the M loop, which is part of the key and lock involved in the inter-protofilament contacts. Importantly, these drugs can modulate the angle between protofilaments, producing microtubules of different diameters. Among the compounds studied, we have found some showing low cytotoxicity and able to induce stabilization without compromising microtubule native structure. This opens the window of new applications for microtubule-stabilizing agents beyond cancer treatment.
PMID: 36462501 10.1016/j.str.2022.11.006
Identification of 1,2,3-triazolium salt-based inhibitors of Leishmania infantum trypanothione disulfide reductase with enhanced antileishmanial potency in cellulo and increased selectivity.
de Lucio H1, Revuelto A2, Carriles AA3, de Castro S2, García-González S2, García-Soriano JC1, Alcón-Calderón M1, Sánchez-Murcia PA4, Hermoso JA3, Gago F5,6, Camarasa MJ2, Jiménez-Ruiz A1, Velázquez S.2
Author information:
1Universidad de Alcalá, Departamento de Biología de Sistemas, 28805 Alcalá de Henares, Madrid, Spain. 2Instituto de Química Médica (IQM, CSIC), Juan de la Cierva 3, 28006 Madrid, Spain. 3Department of Crystallography and Structural Biology, Institute of Physical Chemistry "Rocasolano" (IQFR-CSIC), 28006, Madrid, Spain. 4Division of Physiological Chemistry, Otto Loewi Research Center, Medical University of Graz, Neue Stiftingstalstraße 6/III, 8010, Graz, Austria. 5Universidad de Alcalá, Área de Farmacología, Departamento de Ciencias Biomédicas, 28805 Alcalá de Henares, Madrid, Spain. 6Laboratorio de Modelado Molecular, Unidad asociada al CSIC por el IQM, 28805 Alcalá de Henares, Madrid, Spain.
N-methylation of the triazole moiety present in our recently described triazole-phenyl-thiazole dimerization disruptors of Leishmania infantum trypanothione disulfide reductase (LiTryR) led to a new class of potent inhibitors that target different binding sites on this enzyme. Subtle structural changes among representative library members modified their mechanism of action, switching from models of classical competitive inhibition to time-dependent mixed noncompetitive inhibition. X-ray crystallography and molecular modeling results provided a rationale for this distinct behavior. The remarkable potency and selectivity improvements, particularly against intracellular amastigotes, of the LiTryR dimerization disruptors 4c and 4d reveal that they could be exploited as leishmanicidal agents. Of note, L. infantum promastigotes treated with 4c significantly reduced their low-molecular-weight thiol content, thus providing additional evidence that LiTryR is the main target of this novel compound.
PMID: 36332553 10.1016/j.ejmech.2022.114878
Identification of L. infantum trypanothione synthetase inhibitors with leishmanicidal activity from a (non-biased) in-house chemical library.
Alcón-Calderón M1, de Lucio H1, Revuelto A2, de Castro S2, López-Gutiérrez C1, San-Félix A2, Quesada E2, Gago F3, Camarasa MJ1, Jiménez-Ruiz A1, Velázquez S.2
Author information:
1Universidad de Alcalá, Departamento de Biología de Sistemas, 28805 Alcalá de Henares, Madrid, Spain. 2Instituto de Química Médica (IQM, CSIC), Juan de la Cierva 3, 28006 Madrid, Spain.3Universidad de Alcalá, Área de Farmacología, Laboratorio de Modelado Molecular, Unidad asociada al CSIC por el IQM, Departamento de Ciencias Biomédicas, 28805 Alcalá de Henares, Madrid, Spain.
Redox homeostasis in trypanosomatids is based on the low-molecular-weight trypanothione, an essential dithiol molecule that is synthetized by trypanothione synthetase (TryS) and maintained in its reduced state by trypanothione disulfide reductase (TryR). The fact that both enzymes are indispensable for parasite survival and absent in the mammalian hosts makes them ideal drug targets against leishmaniasis. Although many efforts have been directed to developing TryR inhibitors, much less attention has been focused on TryS. The screening of an in-house library of 144 diverse molecules using two parallel biochemical assays allowed us to detect 13 inhibitors of L. infantum TryS. Compounds 1 and 3 were characterized as competitive inhibitors with Ki values in the low micromolar range and plausible binding modes for them were identified by automated ligand docking against refined protein structures obtained through computational simulation of an entire catalytic cycle. The proposed binding site for both inhibitors overlaps the polyamine site in the enzyme and, additionally, 1 also occupies part of the ATP site. Compound 4 behaves as a mixed hyperbolic inhibitor with a Ki of 0.8 µM. The activity of 5 is clearly dependent on the concentration of the polyamine substrate, but its kinetic behavior is clearly not compatible with a competitive mode of inhibition. Analysis of the activity of the six best inhibitors against intracellular amastigotes identified 5 as the most potent leishmanicidal candidate, with an EC50 value of 0.6 µM and a selectivity index of 35.
PMID: 36075146 10.1016/j.ejmech.2022.114675
A versatile class of 1,4,4-trisubstituted piperidines block coronavirus replication in vitro.
De Castro S1, Stevaert A2, Maldonado M1, Delpal A3, Vandeput J2, Van Loy B2, Eydoux C3, Guillemot JC3, Decroly E3, Gago F4, Canard B3, Camarasa MJ1, Velázquez S1, Naesens L.2
Author information: 1Instituto de Química Médica (IQM, CSIC), E-28006 Madrid, Spain. 2Rega Institute for Medical Research, KU Leuven, B-3000 Leuven, Belgium. 3AFMB, UMR 7257, CNRS, Aix Marseille Université, 13288 Marseille, France. 4Unidad Asociada al IQM-CSIC, Área de Farmacología, Departamento de Ciencias Biomédicas, Universidad de Alcalá, E-28805 Alcalá de Henares, Spain.
There is a clear need for novel antiviral concepts to control SARS-CoV-2 infection. Based on the promising anti-coronavirus activity observed for a class of 1,4,4-trisubstituted piperidines, we here conducted a detailed analysis of the structure-activity relationship of these structurally unique inhibitors. Despite the presence of five points of diversity, the synthesis of an extensive series of analogues was readily achieved by Ugi four-component reaction from commercially available reagents. After evaluating 63 analogues against human coronavirus 229E, four of the best molecules were selected and shown to have micromolar activity against SARS-CoV-2. Since the action point was situated post virus entry and lying at the stage of viral polyprotein processing and the start of RNA synthesis, enzymatic assays were performed with CoV proteins involved in these processes. While no inhibition was observed for SARS-CoV-2 nsp12-nsp7-nsp8 polymerase, nsp14 N7-methyltransferase and nsp16/nsp10 2'-O-methyltransferase, nor the nsp3 papain-like protease, the compounds clearly inhibited the nsp5 main protease (Mpro). Although the inhibitory activity was quite modest, the plausibility of binding to the catalytic site of Mpro was established by in silico studies. Therefore, the 1,4,4-trisubstituted piperidines appear to represent a novel class of non-covalent CoV Mpro inhibitors that warrants further optimization and development.
PMID: 36015168 10.3390/ph15081021
Organotropic dendrons with high potency as HIV-1, HIV-2 and EV-A71 cell entry inhibitors
Martí-Marí O1, Martínez-Gualda B1, Fernández-Barahora I1, Mills A2, Abdelnabi R3, Noppen S3, Neyts J3, Schols D3, Camarasa MJ1, Herranz F1, Gago F2, San-Félix A1
Author information:
1Instituto de Química Médica (IQM-CSIC), c/ Juan de la Cierva 3, E-28006 Madrid, Spain. 2Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.3Department of Microbiology and Immunology, Rega Institute for Medical Research, Laboratory of Virology and Chemotherapy University of Leuven, B-3000, Leuven, Belgium.
We have recently described a novel family of compounds of reduced size and dual anti-HIV and anti-EV71 activity that encompasses tripodal and tetrapodal derivatives. The tripodal prototype, AL-470, has a nitro group at the focal point of the central scaffold and three attached tryptophan residues, each of which bearing an isophthaloyl moiety at the C2 position of the indole ring. A nitro to amino substitution has allowed us now to introduce a chemically addressable functionality to perform further structural modifications consisting of both direct and linker-mediated attachment of several aromatic groups, including the fluorescent dye Alexa Fluor 647 and the antibody-recruiting 2,4-dinitrophenyl motif. Some of the derivatives turned out to be more potent and selective than AL-470 against HIV-1, HIV-2 and EV-A71. The fluorescent probe demonstrated a specific tropism for intestines and lungs, two important niches for the human microbiome in health and disease.
PMID: 35512567 10.1016/j.ejmech.2022.114414
Pyridazino-pyrrolo-quinoxalinium salts as highly potent and selective leishmanicidal agents targeting trypanothione reductase
de Lucio H1, García-Marín J2, García-Marín J2, Sánchez-Alonso P2, García-Soriano JC1, Toro MA1, Vaquero JJ2, Gago F3, Alajarín R2, Jiménez-Ruiz A2
Author information:
1Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 2Departamento de Química, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 3Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
Fifteen pyridazino-pyrrolo-quinoxalinium salts were synthesized and tested for their antiprotozoal activity against Leishmania infantum amastigotes. Eleven of them turned out to be leishmanicidal, with EC50 values in the nanomolar range, and displayed low toxicity against the human THP-1 cell line. Selectivity indices for these compounds range from 10 to more than 1000. Compounds 3b and 3f behave as potent inhibitors of the oxidoreductase activity of the essential enzyme trypanothione disulfide reductase (TryR). Interestingly, binding of 3f is not affected by high trypanothione concentrations, as revealed by the noncompetitive pattern of inhibition observed when tested in the presence of increasing concentrations of this substrate. Furthermore, when analyzed at varying NADPH concentrations, the characteristic pattern of hyperbolic uncompetitive inhibition supports the view that binding of NADPH to TryR is a prerequisite for inhibitor-protein association. Similar to other TryR uncompetitive inhibitors for NADPH, 3f is responsible for TryR-dependent reduction of cytochrome c in a reaction that is typically inhibited by superoxide dismutase.
PMID: 34695777 10.1016/j.ejmech.2021.113915
The structure and flexibility analysis of the Arabidopsis synaptotagmin 1 reveal the basis of its regulation at membrane contact sites.
Benavente JL1, Siliqi D2, Infantes L1, Lagartera L3, Mills A4, Gago F4, Ruiz-López N5, Botella MA5, Sánchez-Barrena MJ1, Albert A.1
Author information:
1 Instituto de Química Física "Rocasolano", Consejo Superior de Investigaciones Científicas (CSIC), E-28006 Madrid, Spain. 2 Istituto di Cristallografia, Consiglio Nazionale delle Ricerche (CNR), Bari, Italy. 3 Instituto de Química Médica (IQM), CSIC, Madrid, Spain. 4 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, Madrid, Spain. 5Departamento de Biología Molecular y Bioquímica. Instituto de Hortofruticultura Subtropical y Mediterránea "La Mayora", Universidad de Málaga-CSIC (IHSM-UMA-CSIC), Universidad de Málaga, Campus de Teatinos, Málaga, Spain.
Non-vesicular lipid transfer at ER and plasma membrane (PM) contact sites (CS) is crucial for the maintenance of membrane lipid homeostasis. Extended synaptotagmins (E-Syts) play a central role in this process as they act as molecular tethers of ER and PM and as lipid transfer proteins between these organelles. E-Syts are proteins constitutively anchored to the ER through an N-terminal hydrophobic segment and bind the PM via a variable number of C-terminal C2 domains. Synaptotagmins (SYTs) are the plant orthologous of E-Syts and regulate the ER-PM communication in response to abiotic stress. Combining different structural and biochemical techniques, we demonstrate that the binding of SYT1 to lipids occurs through a Ca2+-dependent lipid-binding site and by a site for phosphorylated forms of phosphatidylinositol, thus integrating two different molecular signals in response to stress. In addition, we show that SYT1 displays three highly flexible hinge points that provide conformational freedom to facilitate lipid extraction, protein loading, and subsequent transfer between PM and ER.
PMID: 34408000 10.26508/lsa.202101152
Structural and mechanistic insight into DNA bending by antitumour calicheamicins.
Author information:
Department of Biomedical Sciences & "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, E-28805 Alcalá de Henares, Spain.
Among the class of enediyne antibiotics endowed with potent antitumour activities, the calicheamicin derivative known as ozogamicin is selectively targeted to several leukaemia cell types by means of tailor-made immunoconjugates. Binding of these drugs to the DNA minor groove in a sequence-specific fashion eventually causes double-stranded cleavage that results in cell death. Use of calicheamicin ε, an unreactive analogue of calicheamicin γ1I, has demonstrated that these structurally sophisticated molecules inflict bending on certain DNA oligonucleotides of defined sequence to the extent that they increase their circularization ratio upon ligation into multimers. By modelling and simulating several linear and circular DNA constructs containing high-affinity 5'-TCCT-3' and low-affinity 5'-TTGT-3' target sites in the presence and absence of calicheamicin ε, we have shed light into the structural distortions introduced by the drug upon binding to DNA. This new insight not only informs about the direction and magnitude of the DNA curvature but also provides a rationale for an improved understanding of the preferred structural and dynamic features associated with DNA target selection by calicheamicins.
PMID: 34297027 10.1039/D1OB01077H
Small molecule-peptide conjugates as dimerization inhibitors of Leishmania infantum trypanothione disulfide reductase.
Revuelto A1, López-Martín I1, de Lucio H2, García-Soriano JC2, Zanda N1, de Castro S1, Gago F3, Jiménez-Ruiz A2, Camarasa MJ1, Velázquez S.1
Author information: 1Instituto de Química Médica (IQM-CSIC), c/ Juan de la Cierva 3, E-28006 Madrid, Spain. 2Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 3Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
Trypanothione disulfide reductase (TryR) is an essential homodimeric enzyme of trypanosomatid parasites that has been validated as a drug target to fight human infections. Using peptides and peptidomimetics, we previously obtained proof of concept that disrupting protein-protein interactions at the dimer interface of Leishmania infantum TryR (LiTryR) offered an innovative and so far unexploited opportunity for the development of novel antileishmanial agents. Now, we show that linking our previous peptide prototype TRL38 to selected hydrophobic moieties provides a novel series of small-molecule-peptide conjugates that behave as good inhibitors of both LiTryR activity and dimerization.
PMID: 34358115 10.3390/ph14070689
Double arylation of the indole side chain of tri- and tetrapodal tryptophan derivatives renders highly potent HIV-1 and EV-A71 entry inhibitors.
Martí-Marí O1, Martínez-Gualda B1, de la Puente-Secades S1, Mills A2, Quesada E1, Abdelnabi R3, Sun L3, Boonen A3, Noppen S3, Neyts J3, Schols D3, Camarasa MJ1, Gago F2, San-Félix A1
Author information: 1Instituto de Química Médica (IQM-CSIC), c/ Juan de la Cierva 3, E-28006 Madrid, Spain. 2Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 3Laboratory of Virology and Chemotherapy, Department of Microbiology and Immunology, Rega Institute for Medical Research, University of Leuven, B-3000 Leuven, Belgium.
We have recently described a new generation of potent human immunodeficiency virus (HIV) and EV-A71 entry inhibitors. The prototypes contain three or four tryptophan (Trp) residues bearing an isophthalic acid moiety at the C2 position of each side-chain indole ring. This work is now extended by both shifting the position of the isophthalic acid to C7 and synthesizing doubly arylated C2/C7 derivatives. The most potent derivative (50% effective concentration (EC50) HIV-1, 6 nM; EC50 EV-A71, 40 nM), 33 (AL-518), is a C2/C7 doubly arylated tetrapodal compound. Its superior anti-HIV potency with respect to the previous C2-arylated prototype is in consonance with its higher affinity for the viral gp120. 33 (AL-518) showed comparable antiviral activities against X4 and R5 HIV-1 strains and seems to interact with the tip and base of the gp120 V3 loop. Taken together, these findings support the interest in 33 (AL-518) as a useful new prototype for anti-HIV/EV71 drug development.
PMID: 34229438 10.1021/acs.jmedchem.1c00315
[Supporting Information]
On the need to tell apart fraternal twins eEF1A1 and eEF1A2-and their respective outfits.
Author information:
Department of Biomedical Sciences & "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, E-28805 Alcalá de Henares, Spain.
eEF1A1 and eEF1A2 are paralogous proteins whose presence in most normal eukaryotic cells is mutually exclusive and developmentally regulated. Often described in the scientific literature under the collective name eEF1A, which stands for eukaryotic elongation factor 1A, their best known activity (in a monomeric, GTP-bound conformation) is to bind aminoacyl-tRNAs and deliver them to the A-site of the 80S ribosome. However, both eEF1A1 and eEF1A2 are endowed with multitasking abilities (sometimes performed by homo- and heterodimers) and can be located in different subcellular compartments, from the plasma membrane to the nucleus. Given the high sequence identity of these two sister proteins and the large number of post-translational modifications they can undergo, we are often confronted with the dilemma of discerning which is the particular proteoform that is actually responsible for the ascribed biochemical or cellular effects. We argue in this review that acquiring this knowledge is essential to help clarify, in molecular and structural terms, the mechanistic involvement of these two ancestral and abundant G proteins in a variety of fundamental cellular processes other than translation elongation. Of particular importance for this special issue is the fact that several de novo heterozygous missense mutations in the human EEF1A2 gene are associated with a subset of rare but severe neurological syndromes and cardiomyopathies.
PMID: 34203525 10.3390/ijms22136973
Insight into the sequence-specific elements leading to increased DNA bending and ligase-mediated circularization propensity by antitumor trabectedin.
Author information:
Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain.
DNA curvature is the result of a combination of both intrinsic features of the double helix and external distortions introduced by the environment and the binding of proteins or drugs. The propensity of certain double-stranded DNA (dsDNA) sequences to bend is essential in crucial biological processes, such as replication and transcription, in which proteins are known to either recognize noncanonical DNA conformations or promote their formation upon DNA binding. Trabectedin (Yondelis®) is a clinically used antitumor drug which, following covalent bond formation with the 2-amino group of guanine, induces DNA curvature and enhances the circularization ratio, upon DNA ligation, of several dsDNA constructs but not others. By means of unrestrained molecular dynamics simulations using explicitly solvated all-atom models, we rationalize these experimental findings in structural terms and shed light on the crucial, albeit possibly underappreciated, role played by T4 DNA ligase in stabilizing a bent DNA conformation prior to cyclization. Taken together, our results expand our current understanding on how DNA shape modification by trabectedin may affect both the sequence-specific recognition by transcription factors to promoter sites and RNA polymerase II binding.
PMID: 34105031 10.1007/s10822-021-00396-4
[Supplementary Material]
Structural landscape of the transition from an ssDNA dumbbell plus its complementary hairpin to a dsDNA microcircle via a kissing loop intermediate
Author information:
1 Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain.
The experimental construction of a double-stranded DNA microcircle of only 42 base pairs entailed a great deal of ingenuity and hard work. However, figuring out the three-dimensional structures of intermediates and the final product can be particularly baffling. Using a combination of model building and unrestrained molecular dynamics simulations in explicit solvent we have characterized the different DNA structures involved along the process. Our 3D models of the singlestranded DNA molecules provide atomic insight into the recognition event that must take place for the DNA bases in the cohesive tail of the hairpin to pair with their complementary bases in the singlestranded loops of the dumbbell. We propose that a kissing loop involving six base pairs makes up the core of the nascent dsDNA microcircle. We also suggest a feasible pathway for the hybridization of
the remaining complementary bases and characterize the final covalently closed dsDNA microcircle as possessing two well-defined U-turns. Additional models of the pre-ligation complex of T4 DNA ligase with the DNA dumbbell and the post-ligation pre-release complex involving the same enzyme and the covalently closed DNA microcircle are shown to be compatible with enzyme recognition and gap ligation.
PMID: 34069399 10.3390/molecules26103017
Efficient dimerization disruption of Leishmania infantum trypanothione reductase by triazole-phenyl-thiazoles.
Revuelto A1, de Lucio H2, García-Soriano JC2, Sánchez-Murcia PA3, Gago F3, Jiménez-Ruiz A2, Camarasa MJ1, Velázquez S.1
Author information: 1Instituto de Química Médica (IQM-CSIC), c/ Juan de la Cierva 3, E-28006 Madrid, Spain. 2Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 3Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
Inhibition of Leishmania infantum trypanothione disulfide reductase (LiTryR) by disruption of its homodimeric interface has proved to be an alternative and unexploited strategy in the search for novel antileishmanial agents. Proof of concept was first obtained by peptides and peptidomimetics. Building on previously reported dimerization disruptors containing an imidazole-phenyl-thiazole scaffold, we now report a new 1,2,3-triazole-based chemotype that yields noncompetitive, slow-binding inhibitors of LiTryR. Several compounds bearing (poly)aromatic substituents dramatically improve the ability to disrupt LiTryR dimerization relative to reference imidazoles. Molecular modeling studies identified an almost unexplored hydrophobic region at the interfacial domain as the putative binding site for these compounds. A subsequent structure-based design led to a symmetrical triazole analogue that displayed even more potent inhibitory activity over LiTryR and enhanced leishmanicidal activity. Remarkably, several of these novel triazole-bearing compounds were able to kill both extracellular and intracellular parasites in cell cultures.
PMID: 33945281 10.1021/acs.jmedchem.1c00206
[Supporting Information]
Multivalent tryptophan- and tyrosine-containing [60]fullerene hexa-adducts as dual HIV and Enterovirus A71 entry inhibitors.
Ruiz-Santaquiteria M1, Illescas BM1, Abdelnabi R2, Boonen A2, Mills A3, Martí-Marí O1, Noppen S2, Neyts J2, Schols D2, Gago F3, San-Félix A4, Camarasa MJ4, Martín N1.
1University Complutense, Organic Chemistry, Faculty of Chemistry, 28040 Madrid, Spain.
2Laboratory of Virology and Chemotherapy, Rega Institute for Medical Research, Katholieke Universiteit Leuven, Belgium.
3Department of Biomedical Sciences and "U.A. IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, E-28805 Madrid, Spain.
4Instituto de Química Médica, Consejo Superior de Investigaciones Cientificas, Madrid, Spain.
Unprecedented 3D hexa-adducts of [60]fullerene peripherally decorated with twelve tryptophan (Trp) or tyrosine (Tyr) residues have been synthesized. Studies on the antiviral activity of these novel compounds against HIV and EV71 reveal that they are much more potent against HIV and equally active against EV71 than the previously described dendrimer prototypes AL-385 and AL-463, which possess the same number of Trp/Tyr residues on the periphery but attached to a smaller and more flexible pentaerythritol core. These results demonstrate the relevance of the globular 3D presentation of the peripheral groups (Trp/Tyr) as well as the length of the spacer connecting them to the central core to interact with the viral envelopes, particularly in the case of HIV, and support the hypothesis that [60]fullerene can be an alternative and attractive biocompatible carbon-based scaffold for this type of highly symmetrical dendrimers. In addition, the functionalized fullerenes here described, which display twelve peripheral negatively charged indole moieties on their globular surface, define a new and versatile class of compounds with a promising potential in biomedical applications.
© 2021 Wiley-VCH GmbH.
PMID: 33851758 10.1002/chem.202101098
[Supporting Information]
Structural cues for understanding eEF1A2 moonlighting.
Carriles AA1, Mills A2, Muñoz-Alonso MJ3, Domínguez JM3, Gutiérrez D4, Hermoso JA1,Gago F2
1Instituto de Química Física Rocasolano, Consejo Superior de Investigaciones Científicas (CSIC), E-28006 Madrid, Spain.
2Department of Biomedical Sciences and "U.A. IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, E-28805 Madrid, Spain.
3Cell Biology, PharmaMar SA, Colmenar Viejo, E-28770 Madrid, Spain.
4Proteomics Unit, Facultad de Farmacia, Universidad Complutense, E-28040 Madrid, Spain.
Spontaneous mutations in the EEF1A2 gene cause epilepsy and severe neurological disabilities in children. The crystal structure of eEF1A2 protein purified from rabbit skeletal muscle reveals a post-translationally modified dimer that informs about sites of interaction with numerous binding partners, including itself, and maps these mutations onto the dimer and tetramer interfaces. The spatial locations of the side chain carboxylates of Glu301 and Glu374, to which phosphatidylethanolamine is uniquely attached via an amide bond, define the anchoring points of eEF1A2 to cellular membranes and interorganellar membrane contact sites. Additional bioinformatic and molecular modelling results provide novel structural insight into the demonstrated binding of eEF1A2 to SH3 domains, the common MAPK docking groove, filamentous actin, and phosphatidylinositol-4 kinase IIIβ. In this new light, the role of eEF1A2 as an ancient, multifaceted, and articulated protein at the crossroads of autophagy, oncogenesis and viral replication appears very distant from the "canonical" one of delivering aminoacylated tRNAs to the ribosome that has dominated the scene and much of the thinking for many decades.
© 2020 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
PMID: 32875694 10.1002/cbic.202000516
[Supporting Information] [Movie S1: Simulated conformational transition between GDP-bound dimeric eEF1A2 and the GTP-bound monomeric eEF1A2 conformation capable of binding aminoacyl-tRNA and delivering it to the ribosome.] [Movie S2: In crystallo MD simulation of a fully solvated eEF1A2 dimer:dimer ensemble.] [Movie S3: MD simulation of two fully solvated eEF1A2 dimers as found in the crystal lattice.] [Movie S4: MD simulation of a fully solvated eEF1A2 monomer in the GTP conformation.]
| |
Structure reported in this article: 6RA9 |
Pseudoirreversible slow-binding inhibition of trypanothione reductase by a protein-protein interaction disruptor.
de Lucio H1, Toro MA2, Camarasa MJ3, Velázquez S3, Gago F4, Jiménez-Ruiz A1.
1Área de Bioquímica y Biología Molecular, Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
2Centro Nacional de Secuenciación Genómica-CNSG, Universidad de Antioquia, Medellin, Antioquia, Colombia.
3Instituto de Química Médica (IQM-CSIC), E-28006 Madrid, Spain.
4Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
BACKGROUND AND PURPOSE: Peptide P4 was previously described as a dimerization disruptor of trypanothione reductase (TryR), a homodimeric enzyme essential for survival of trypanosomatids. Determination of the true inhibitory constant (Ki) for P4 was not achieved then because, even under conditions in which substrate concentration was kept constant, reaction rates continuously decreased with time. The aim of this study was to find a suitable kinetic model that could allow characterization of the complex pattern of TryR inhibition caused by P4.
EXPERIMENTAL APPROACH: After proving the slow-binding and pseudoirreversible activity of P4 against Leishmania infantum TryR (Li-TryR), analysis of the curvatures of the reaction progress curves at different inhibitor concentrations allowed us to define the apparent inhibitory constants (Kiapp) at five different substrate concentrations. Analysis of the changes in Kiapp values allowed precise definition of the type of inhibition.
KEY RESULTS: Li-TryR inhibition by P4 requires two sequential steps that involve rapid generation of a reversible enzyme-inhibitor complex followed by a pseudoirreversible slow inactivation of the enzyme. Recovery of enzyme activity after inhibitor dissociation is barely detectable. P4 is a noncompetitive pseudoirreversible inhibitor of Li-TryR that displays an overall inhibition constant (Ki*) smaller than 0.02 µM.
CONCLUSIONS AND IMPLICATIONS: Li-TryR dimer disruption by peptide P4 is a pseudoirreversible time-dependent process which is noncompetitive with respect to the oxidized trypanothione (TS2) substrate. Therefore, unlike reversible Li-TryR competitive inhibitors, enzyme inhibition by P4 is not affected by the TS2 accumulation observed during oxidant processes such as the oxidative burst in host macrophages.
PMID: 32888319 10.1111/bph.15250
Distinct binding of cetirizine enantiomers to human serum albumin and the human histamine receptor H1.
Perona A1,2, Ros MP1, Mills A3, Morreale A4, Gago F.3
Author information:
1Universidad Europea de Madrid, Urbanización El Bosque, c/ Tajo s/n, 28670, Villaviciosa de Odón, Madrid, Spain. 2Departamento de Química en Ciencias Farmacéuticas, Facultad de Farmacia, Ciudad Universitaria, Plaza de Ramón y Cajal, 28040, Madrid, Spain. 3Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, 28805,
Alcalá de Henares, Madrid, Spain. 4Repsol Technology Lab, c/ Agustín de Betancourt s/n, 28935, Móstoles, Madrid, Spain.
Cetirizine, a major metabolite of hydroxyzine, became a marketed second-generation H1 antihistamine that is orally active and has a rapid onset of action, long duration of effects and a very good safety record at recommended doses. The approved drug is a racemic mixture of (S)-cetirizine and (R)-cetirizine, the latter being the levorotary enantiomer that also exists in the market as a third-generation, non-sedating and highly selective antihistamine. Both enantiomers bind tightly to the human histamine H1 receptor (hH1R) and behave as inverse agonists but the affinity and residence time of (R)-cetirizine are greater than those of (S)-cetirizine. In blood plasma, cetirizine exists in the zwitterionic form and more than 90% of the circulating drug is bound to human serum albumin (HSA), which acts as an inactive reservoir. Independent X-ray crystallographic work has solved the structure of the hH1R:doxepin complex and has identified two drug-binding sites for cetirizine on equine serum albumin (ESA). Given this background, we decided to model a membrane-embedded hH1 in complex with either (R)- or (S)-cetirizine and also the complexes of both ESA and HSA with these two enantiomeric drugs to analyze possible differences in binding modes between enantiomers and also among targets. The ensuing molecular dynamics simulations in explicit solvent and additional computational chemistry calculations provided structural and energetic information about all of these complexes that is normally beyond current experimental possibilities. Overall, we found very good agreement between our binding energy estimates and extant biochemical and pharmacological evidence. A much higher degree of solvent exposure in the cetirizine-binding site(s) of HSA and ESA relative to the more occluded orthosteric binding site in hH1R is translated into larger positional fluctuations and considerably lower affinities for these two nonspecific targets. Whereas it is demonstrated that the two known pockets in ESA provide enough stability for cetirizine binding, only one such site does so in HSA due to a number of amino acid replacements. At the histamine-binding site in hH1R, the distinct interactions established between the phenyl and chlorophenyl moieties of the two enantiomers with the amino acids lining up the pocket and between their free carboxylates and Lys179 in the second extracellular loop account for the improved pharmacological profile of (R)-cetirizine.
PMID: 32572668 10.1007/s10822-020-00328-8
[Electronic Supplementary Material]
[Electronic Supplementary Material (PDF)]
N-Ribosyltransferase from Archaeoglobus veneficus: A novel halotolerant and thermostable biocatalyst for the synthesis of purine ribonucleoside analogs.
Acosta J1, del Arco1, Pisabarro V1, Gago F2, Fernández-Lucas J.1,3
1Applied Biotechnology Group, European University of Madrid, Villaviciosa de Odón, Spain. 2Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, Spain. 3Grupo de Investigación en Ciencias Naturales y Exactas, GICNEX, Universidad de la Costa, CUC, Barranquilla, Colombia
Nucleoside-2'-deoxyribosyl-transferases (NDTs) catalyze a transglycosylation reaction consisting of the exchange of the 2'-deoxyribose moiety between a purine and/or pyrimidine nucleoside and a purine and/or pyrimidine base. Because NDTs are highly specific for 2'-deoxyribonucleosides they generally display poor activity on modified C2' and C3' nucleosides and this limitation hampers their applicability as biocatalysts for the synthesis of modified nucleosides. We now report the production and purification of a novel NDT from Archaeoglobus veneficus that is endowed with native ribosyltransferase activity and hence it is more properly classified as an N-ribosyltransferase (AvNRT). Biophysical and biochemical characterization revealed that AvNRT is a homotetramer that displays maximum activity at 80°C and pH 6 and shows remarkably high stability at high temperatures (60-80 °C). In addition, the activity of AvNRT was found to increase up to 2-fold in 4 M NaCl aqueous solution and to be retained in the presence of several water-miscible organic solvents. For completeness, and as a proof of concept for possible industrial applications, this thermophilic and halotolerant biocatalyst was successfully employed in the synthesis of different purine ribonucleoside analogs.
PMID: 32612982 10.3389/fbioe.2020.00593
Hakin-1, a new specific small-molecule inhibitor for the E3 ubiquitin-ligase Hakai, inhibits carcinoma growth and progression
Martínez-Iglesias Oa, Casas-Pais Aa, Castosa Ta, Díaz Díaz Aa, Roca-Lema Da, Concha Ab, Cortés Ac, Gago Fd, Figueroa A.a
Author information:
a Epithelial Plasticity and Metastasis Group, Instituto de Investigación Biomédica de A Coruña (INIBIC), Complexo Hospitalario Universitario de A Coruña (CHUAC), Sergas, Universidade da Coruña (UDC), 15006 A Coruña, Spain. b Pathology Department and A Coruña Biobank from Instituto de Investigación Biomédica de A Coruña (INIBIC), Complexo Hospitalario Universitario de A Coruña (CHUAC), Sergas, Universidade da Coruña (UDC), 15006 A Coruña, Spain. c Computational Chemistry-UK, RD Platform Technology & Science, GSK Medicines Research Centre, Hertfordshire SG12 0DP, UK dArea of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá de Henares, 28805 Alcalá de Henares, Spain
The requirement of the E3 ubiquitin-ligase Hakai for the ubiquitination and subsequent degradation of E-cadherin has been associated with enhanced epithelial-to-mesenchymal transition (EMT), tumour progression and carcinoma metastasis. To date, most of the reported EMT-related inhibitors were not developed for anti-EMT purposes, but indirectly affect EMT. On the other hand, E3 ubiquitin-ligase enzymes have recently emerged as promising therapeutic targets, as their specific inhibition would prevent wider side effects. Given this background, a virtual screening was performed to identify novel specific inhibitors of Hakai, targeted against its phosphotyrosine-binding pocket, where phosphorylated-E-cadherin specifically binds. We selected a candidate inhibitor, Hakin-1, which showed an important effect on Hakai-induced ubiquitination. Hakin-1 also inhibited carcinoma growth and tumour progression both in vitro, in colorectal cancer cell lines, and in vivo, in a tumour xenograft mouse model, without apparent systemic toxicity in mice. Our results show for the first time that a small molecule putatively targeting the E3 ubiquitin-ligase Hakai inhibits Hakai-dependent ubiquitination of E-cadherin, having an impact on the EMT process. This represents an important step forward in a future development of an effective therapeutic drug to prevent or inhibit carcinoma tumour progression.
PMID: 32456234 10.3390/cancers12051340
N-benzyl 4,4-disubstituted piperidines as a potent class of influenza H1N1 virus inhibitors showing a novel mechanism of hemagglutinin fusion peptide interaction
de Castro Sa, Ginex Tb, Vanderlinden Ec, Laporte Mc, Stevaert Ac, Cumella Ja, Gago Fd, Camarasa MJa, Luque FJe, Naesens Lf,Velázquez S.a
Author information:
a Instituto de Química Médica, Consejo Superior de Investigaciones Científicas (IQM-CSIC), E-28006 Madrid, Spain. b Departament de Nutrició, Ciències de l'Alimentació I Gastronomia, Institut de Biomedicina (IBUB) and Institut de Química Teòrica i Computacional (IQTCUB), Facultat de Farmàcia i Ciències de l'Alimentació, Universitat de Barcelona, Santa Coloma de Gramenet, Spain. c Rega Institute for Medical Research, KU Leuven, University of Leuven, B-3000, Belgium. dÁrea de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. eDepartament de Nutrició, Ciències de l'Alimentació I Gastronomia, Institut de Biomedicina (IBUB) and Institut de Química Teòrica i Computacional (IQTCUB), Facultat de Farmàcia i Ciències de l'Alimentació, Universitat de Barcelona, Santa Coloma de Gramenet, Spain. Electronic address: fjluque@ub.edu. fRega Institute for Medical Research, KU Leuven, University of Leuven, B-3000, Belgium. Electronic address: lieve.naesens@kuleuven.be.
The influenza virus hemagglutinin (HA) is an attractive target for antiviral therapy due to its essential role in mediating virus entry into the host cell. We here report the identification of a class of N-benzyl-4,4,-disubstituted piperidines as influenza A virus fusion inhibitors with specific activity against the H1N1 subtype. Using the highly efficient one-step Ugi four-component reaction, diverse library of piperidine-based analogues was synthesized and evaluated to explore the structure-activity relationships (SAR). Mechanistic studies, including resistance selection with the most active compound (2) demonstrated that it acts as an inhibitor of the low pH-induced HA-mediated membrane fusion process. Computational studies identified an as yet unrecognized fusion inhibitor binding site, which is located at the bottom of the HA2 stem in close proximity to the fusion peptide. A direct ?-stacking interaction between the N-benzylpiperidine moiety of 2 and F9HA2 of the fusion peptide, reinforced with an additional ?-stacking interaction with Y119HA2, and a salt bridge of the protonated piperidine nitrogen with E120HA2, were identified as important interactions to mediate ligand binding. This site rationalized the observed SAR and provided a structural explanation for the H1N1-specific activity of our inhibitors. Furthermore, the HA1-S326V mutation resulting in resistance to 2 is close to the proposed new binding pocket. Our findings point to the N-benzyl-4,4,-disubstituted piperidines as an interesting class of influenza virus inhibitors, representing the first example of fusion peptide binders with great potential for anti-influenza drug development.
PMID: 32220685 10.1016/j.ejmech.2020.112223
Peptides mimicking the β7/β8 loop of HIV-1 reverse transcriptase p51 as "hotspot-targeted" dimerization inhibitors
Sánchez-Murcia PA1,6, de Castro S1, García-Aparicio C1, Jiménez MA2 Corona A3, Tramontano E3, Sluis-Cremer N4, Menéndez-Arias L5, Velázquez S1, Gago F6, Camarasa MJ.1
Author information:
1 Instituto de Química Médica (IQM-CSIC), E-28006 Madrid, Spain. 2 Instituto de Química-Física Rocasolano (IQFR, CSIC), Serrano 119, E-28006 Madrid, Spain. 3 University of Cagliari, Department of Life and Environmental Sciences, Cittadella Universitaria di Monserrato, 09042 Monserrato, Cagliari, Italy. 4 University of Pittsburgh School of Medicine, Division of Infectious Diseases, Pittsburgh, Pennsylvania 15261, United States. 5 Centro de Biología Molecular "Severo Ochoa" (CBMSO, CSIC & Universidad Autónoma de Madrid), Nicolás Cabrera 1, Campus de Cantoblanco, 28049 Madrid, Spain.6 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
A conformationally constrained short peptide designed to target a protein-protein interaction hotspot in HIV-1 reverse transcriptase (RT) disrupts p66-p51 interactions and paves the way to the development of novel RT dimerization inhibitors.
PMID: 32435389 10.1021/acsmedchemlett.9b00623
[Supporting Info]
Recognition and activation of the plant AKT1 potassium channel by the kinase CIPK23
Sánchez-Barrena MJ1, Chaves-Sanjuan A2, Raddatz N3, Mendoza I3, Cortés A4, Gago F4, González-Rubio JM1, Benavente JL1, Quintero FJ2, Pardo JM2, Albert A.1
Author information:
1 Instituto de Química Física (IQFR), Consejo Superior de Investigaciones Científicas (CSIC), E-28006 Madrid, Spain. 2 University of Milan. 3 Instituto de Bioquimica Vegetal y Fotosintesis (IBVF), Consejo Superior de Investigaciones Científicas (CSIC) and University of Seville, E-41902 Sevilla, Spain. 4 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
Plant growth largely depends on the maintenance of adequate intracellular levels of potassium (K+). The families of 10 Calcineurin B-Like (CBL) calcium sensors and 26 CBL-Interacting Protein Kinases (CIPKs) of Arabidopsis thaliana decode the calcium signals elicited by environmental inputs to regulate different ion channels and transporters involved in the control of K+ fluxes by phosphorylation-dependent and -independent events. However, the detailed molecular mechanisms governing target specificity require investigation. Here, we show that the physical interaction between CIPK23 and the non-canonical ankyrin domain in the cytosolic side of the inward-rectifier K+ channel AKT1 regulates kinase docking and channel activation. Point mutations on this domain specifically alter binding to CIPK23, enhancing or impairing the ability of CIPK23 to regulate channel activity. Our data demonstrate the relevance of this protein-protein interaction that contributes to the formation of a complex between CIPK23/CBL1 and AKT1 in the membrane for the proper regulation of K+ transport.
PMID: 32015077 10.1104/pp.19.01084
Tuning melatonin receptor subtype selectivity in oxadiazolone-based analogues: Discovery of QR2 ligands and NRF2 activators with neurogenic properties
Herrera-Arozamena C1, Estrada-Valencia M1, Pérez C1, Lagartera L1, Morales-García J2, Pérez-Castillo A2, Franco-González JF4, Michalska P4, Duarte P4, León R4, López M1, Mills A3, Gago F3, Rodríguez-Franco MI.1
Author information:
1 Instituto de Química Médica, Consejo Superior de Investigaciones Científicas (IQM-CSIC), E-28006 Madrid, Spain. 2 Instituto de Investigaciones Biomédicas (CSIC-UAM), E-28029 Madrid, Spain, and Centro de Investigación Biomédica en Red Sobre Enfermedades Neurodegenerativas (CIBERNED). 3 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 4 Instituto Teófilo Hernando de I+D del Medicamento, Departamento de Farmacología y Terapéutica, Facultad de Medicina, Universidad Autónoma de Madrid, E-28029 Madrid, Spain.
New multi-target indole and naphthalene derivatives containing the oxadiazolone scaffold as a bioisostere of the melatonin acetamido group have been developed. The novel compounds were characterized at melatonin receptors MT1R and MT2R, quinone reductase 2 (QR2), lipoxygenase-5 (LOX-5), and monoamine oxidases (MAO-A and MAO-B), and also as radical scavengers. We found that selectivity within the oxadiazolone series can be modulated by modifying the side chain functionality and co-planarity with the indole or naphthalene ring. In phenotypic assays, several oxadiazolone-based derivatives induced signalling mediated by the transcription factor NRF2 and promoted the maturation of neural stem-cells into a neuronal phenotype. Activation of NRF2 could be due to the binding of indole derivatives to KEAP1, as deduced from surface plasmon resonance (SPR) experiments. Molecular modelling studies using the crystal structures of QR2 and the KEAP1 Kelch-domain, as well as the recently described X-ray free-electron laser (XFEL) structures of chimeric MT1R and MT2R, provided a rationale for the experimental data and afforded valuable insights for future drug design endeavours.
PMID: 32018096 10.1016/j.ejmech.2020.112090
[Supplementary data]
Atomistic insight into sequence-directed DNA bending and minicircle formation propensity in the absence and presence of phased A-tracts.
Author information:
Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain.
Bending of double-stranded (ds) DNA plays a crucial role in many important biological processes and is relevant for nanotechnological applications. Among all the elements that have been studied in relation to dsDNA bending, A-tracts stand out as one of the most controversial. The "ApA wedge" theory was disproved when a series of linear polynucleotides containing phased 5'-A4T4-3' or 5'-T4A4-3' runs were shown to be bent or straight, respectively, and crystallographic evidence revealed that A-tracts are unbent. Furthermore, some of the smallest dsDNA minicircles described to date (∼100 bp in size) lack A-tracts and are subjected to varying levels of torsional stress. Representative DNA sequences from this experimental background were modeled in atomic detail and their dynamic behavior was simulated over hundreds of nanoseconds using the AMBER force field ParmBSC1. Subsequent analysis of the resulting trajectories allowed us to (i) unambiguously establish the location of the bends in all cases; (ii) identify the structural elements that are directly responsible for the macroscopically detected curvature; and (iii) reveal the importance not only of coherently summing the effects of the bending elements when they are in synchrony with the natural repeat of the helix (i.e. separated by an integral number of helical turns) but also when alternated with a half-integral separation of opposite effects. We conclude that the major determinant of the macroscopically observed bending is the proper grouping and phasing of the positive roll imposed by pyrimidine-purine (YR) steps and the negative or null roll characteristic of RY steps and A-tracts, respectively. This conclusion is in very good agreement with the structural parameters experimentally derived for much smaller DNA molecules either alone or as found in DNA-protein complexes. We expect that this work will pave the way for future studies on drug-induced DNA bending, DNA shape readout by transcription factors, structure of circular extrachromosomal DNA, and custom design of curved DNA origami scaffolds.
PMID: 31950463 10.1007/s10822-020-00288-z
[Supplementary Material]
A supramolecular stabilizer of the 14-3-3ζ/ERα protein-protein interaction with a synergistic mode of action.
Gigante A1, Sijbesma E1, Sánchez-Murcia PA2, Hu X1, Bier D1, Bäcker S1, Knauer S1, Gago F3, Ottmann C1, Schmuck C.1
1Technische Universiteit Eindhoven, 5600 MB Eindhoven, The Netherlands. 2Institute of Theoretical Chemistry, Faculty of Chemistry, Währinger Str. 17, A-1090 University of Vienna, Vienna, Austria. 3Departamento de Ciencias Biomédicas, Universidad de Alcalá, Alcalá de Henares, E-28805 Madrid, Spain.
We report on a stabilizer of the interaction between 14-3-3ζ and the Estrogen Receptor α (ERα). ERα is a driver in the majority of breast cancers and 14-3-3 proteins are negative regulators of this Nuclear Receptor, making the stabilization of this protein-protein interaction (PPI) an interesting strategy. The stabilizer 1 consists of three symmetric peptidic arms containing an arginine mimetic, previously described as the GCP motif. 1 stabilizes the 14-3-3ζ/ERα interaction synergistically with the natural product Fusicoccin A, and was thus hypothesized to bind to a different site. This is supported by a computational analysis of 1 binding to the binary complex of 14-3-3 and an ERα-derived phosphopeptide. Furthermore, 1 shows selectivity towards the 14-3-3ζ/ERα interaction over other 14-3-3 client-derived phosphomotifs. These data provide a solid support of a new binding mode for a supramolecular 14-3-3ζ/ERα PPI stabilizer.
PMID: 31814236 10.1002/anie.201914517
Generation of endoplasmic reticulum stress and inhibition of autophagy by plitidepsin induces proteotoxic apoptosis in cancer cells.
Losada A1, Berlanga JJ2, Molina-Guijarro JM1, Jiménez-Ruiz A3, Gago F4, Avilés P1, de Haro C2, Martínez-Leal JF.1
1Departamento de Investigación y Desarrollo, Pharma Mar SA, Colmenar Viejo, Madrid E-28770, Spain. 2Departamento de Biología Molecular, CBMSO, Cantoblanco, Madrid E-28049, Spain. 3Departamento de Biología de Sistemas, Universidad Alcalá de Henares, Alcalá de Henares, Madrid E-28805, Spain. 4Departamento de Ciencias Biomédicas, Universidad Alcalá de Henares, Alcalá de Henares, Madrid E-28805, Spain.
Plitidepsin (PLD, Aplidin®), a cyclic depsipeptide originally isolated from the marine tunicate Aplidium albicans, has been recently approved by Australian regulatory authorities for the treatment of multiple myeloma patients. Plitidepsin binds to eEF1A2 and induces oxidative stress, Rac1 activation and JNK1 phosphorylation, triggering a rapid apoptotic program in tumor cells. Since oxidative stress is one of the known sources of endoplasmic reticulum stress, we investigated whether PLD was inducing a bona fide ER stress in HeLa cells and whether this process was essential in the mechanism of action of the compound. Indeed, PLD activated an ER stress-induced unfolded protein response (UPR), including the alternative splicing of XBP1, the proteolytic processing of ATF6 and the phosphorylation of eIF2α and JNK. Interestingly, though PLD induced a strong phosphorylation of eIF2α in all the analyzed cell lines, it did not elicit an increased expression of ATF4 and CHOP, a transcription factor involved in launching UPR-mediated apoptosis. On the contrary, a clear reduction of CHOP protein levels was observed after PLD treatment, most probably due to both the lack of transactivation by ATF4 and its rapid degradation by the ubiquitin/proteasome machinery. Using fibroblasts devoid of each one of the four possible kinases involved in eIF2α phosphorylation, we observed that only PKR was involved in the response to PLD treatment and, accordingly, PKR-/- fibroblasts are shown to be resistant to the apoptogenic activity of the compound. Furthermore, eIF2α phosphorylation itself was shown to be irrelevant for the induction of cell death by PLD. Instead, we reveal that PLD induces an increase in the levels of misfolded proteins while simultaneously inhibiting the autophagic flux. These two effects combined prevent PLD-treated cells from reducing proteotoxic stress and lead to apoptosis. Other anti-myeloma drugs like bortezomib, which target the proteasome, also inhibit the degradation of misfolded proteins through alternate pathways and a synergistic anticancer effect of the PLD plus bortezomib combination has been previously disclosed. The present results extend this synergy to in vivo experiments and provide a mechanistic rationale for this synergy.
PMID: 31812675 10.1016/j.bcp.2019.113744
Scaffold simplification strategy leads to a novel generation of dual human immunodeficiency virus and enterovirus-A71 entry inhibitors.
Martínez-Gualda B1, Sun L2, Martí-Marí O1, Noppen S2, Abdelnabi R2, Bator CM3, Quesada E1, Delang L2, Mirabelli C2, Lee H3, Schols D2, Neyts J2, Hafenstein S3, Camarasa MJ1, Gago F5, San-Félix A1.
Author information: 1Instituto de Química Médica (IQM-CSIC) , 28006 Madrid , Spain. 2Department of Microbiology and Immunology, Rega Institute for Medical Research, Laboratory of Virology and Chemotherapy, University of Leuven, B-3000 Leuven, Belgium. 3Department of Biochemistry and Molecular Biology, Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, 16802 State College, Pennsylvania, United States. 4Department of Medicine , The Pennsylvania State University College of Medicine, 17033 Hershey, Pennsylvania, United States. 5Departamento de Ciencias Biomédicas y Unidad Asociada IQM-UAH, Universidad de Alcalá, Alcalá de Henares, E-28805 Madrid, Spain.
Currently there are only three FDA-approved drugs that inhibit HIV entry-fusion into host cells. The situation is even worse for enterovirus EV71 infection for which no antiviral therapies are available. We describe here the discovery of potent entry dual inhibitors of HIV and EV71. These compounds contain in their structure three or four tryptophan (Trp) residues linked to a central scaffold. Critical for anti-HIV/EV71 activity is the presence of extra phenyl rings, bearing one or two carboxylates, at the C2 position of the indole ring of each Trp residue. The most potent derivatives, 22 and 30, inhibit early steps of the replicative cycles of HIV-1 and EV-A71 by interacting with their respective viral surfaces (glycoprotein gp120 of HIV and 5-fold axis of the EV-A71 capsid). The high potency, low toxicity, facile chemical synthesis and great opportunities for chemical optimization make them useful prototypes for future medicinal chemistry studies.
PMID: 31809045 10.1021/acs.jmedchem.9b01737
Differential phenotypic expression of a novel PDHA1 mutation in a female monozygotic twin pair.
Horga A1,2, Woodward CE3, Mills A4, Pareés I5, Hargreaves IP6, Brown RM7, Bugiardini E1, Brooks T8, Manole A1, Remzova E6, Rahman S9, Reilly MM1, Houlden H1, Sweeney MG3, Brown GK7, Polke JM3, Gago F4, Parton MJ1, Pitceathly RDS10, Hanna MG.11
1Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology and The National Hospital for Neurology and Neurosurgery, London, UK. 2Neuromuscular Diseases Unit, Department of Neurology, Hospital Clínico San Carlos, IdISSC, Madrid, Spain. 3Neurogenetics Unit, The National Hospital for Neurology and Neurosurgery, London, UK. 4Area of Pharmacology, Department of Biomedical Sciences, School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, Spain. 5Sobell Department of Motor Neuroscience and Movement Disorders, UCL Queen Square Institute of Neurology, London, UK. 6Neurometabolic Unit, The National Hospital for Neurology and Neurosurgery, London, UK. 7Oxford Medical Genetics Laboratories, The Churchill Hospital, Oxford University Hospitals NHS Foundation Trust, Oxford, UK. 8UCL Genomics, UCL Great Ormond Street Institute of Child Health, University College London, London, UK. 9Metabolic Unit, Great Ormond Street Hospital for Children NHS Foundation Trust and UCL Great Ormond Street Institute of Child Health, London, UK. 10Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology and The National Hospital for Neurology and Neurosurgery, London, UK. 11Department of Neuromuscular Diseases, UCL Queen Square Institute of Neurology and The National Hospital for Neurology and Neurosurgery, London, UK.
Pyruvate dehydrogenase complex (PDC) deficiency caused by mutations in the X-linked PDHA1 gene has a broad clinical presentation, and the pattern of X-chromosome inactivation has been proposed as a major factor contributing to its variable expressivity in heterozygous females. Here, we report the first set of monozygotic twin females with PDC deficiency, caused by a novel, de novo heterozygous missense mutation in exon 11 of PDHA1 (NM_000284.3: c.1100A>T). Both twins presented in infancy with a similar clinical phenotype including developmental delay, episodes of hypotonia or encephalopathy, epilepsy, and slowly progressive motor impairment due to pyramidal, extrapyramidal, and cerebellar involvement. However, they exhibited clear differences in disease severity that correlated well with residual PDC activities (approximately 60% and 20% of mean control values, respectively) and levels of immunoreactive E1α subunit in cultured skin fibroblasts. To address whether the observed clinical and biochemical differences could be explained by the pattern of X-chromosome inactivation, we undertook an androgen receptor assay in peripheral blood. In the less severely affected twin, a significant bias in the relative activity of the two X chromosomes with a ratio of approximately 75:25 was detected, while the ratio was close to 50:50 in the other twin. Although it may be difficult to extrapolate these results to other tissues, our observation provides further support to the hypothesis that the pattern of X-chromosome inactivation may influence the phenotypic expression of the same mutation in heterozygous females and broadens the clinical and genetic spectrum of PDC deficiency.
PMID: 31673819 10.1007/s00439-019-02075-9
[Supplementary Material]
Reaction mechanism of nucleoside 2'-deoxyribosyltransferases: free-energy landscape supports an oxocarbenium ion as the reaction intermediate.
Del Arco J1, Perona A1, González L2, Fernández-Lucas J3, Gago F4, Sánchez-Murcia PA.2
1Applied Biotechnology Group, European University of Madrid, Villaviciosa de Odón, Spain. 2Institute of Theoretical Chemistry, Faculty of Chemistry, Währinger Str. 17, A-1090 University of Vienna, Vienna, Austria. 3Applied Biotechnology Group, European University of Madrid, Villaviciosa de Odón, Spain and Grupo de Investigación en Ciencias Naturales y Exactas, GICNEX, Universidad de la Costa, CUC, Calle 58 # 55-66, Barranquilla, Colombia. 4Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, Spain.
Insight into the catalytic mechanism of Lactobacillus leichmannii nucleoside 2'-deoxyribosyltransferase (LlNDT) has been gained by calculating a quantum mechanics-molecular mechanics (QM/MM) free-energy landscape of the reaction within the enzyme active site. Our results support an oxocarbenium species as the reaction intermediate and thus an SN1 reaction mechanism in this family of bacterial enzymes. Our mechanistic proposal is validated by comparing experimental kinetic data on the impact of the single amino acid replacements Tyr7, Glu98 and Met125 with Ala, Asp and Ala/norLeu, respectively, and accounts for the specificity shown by this enzyme on a non-natural substrate. This work broadens our understanding of enzymatic C-N bond cleavage and C-N bond formation.
PMID: 31397456 10.1039/c9ob01315f
[Supplementary Information (txt)]
[Supplementary Information (zip)]
[Supplementary Information (pdf)]
Structure-guided tuning of a selectivity switch towards ribonucleosides in Trypanosoma brucei purine nucleoside 2' deoxyribosyltransferase.
Del Arco J1, Mills A2, Gago F2, Fernández-Lucas, J.1
1 Applied Biotechnology Group, Universidad Europea de Madrid, Urbanización El Bosque, c/ Tajo, s/n, Villaviciosa de Odón, 28670 Madrid, Spain.
2 Department of Biomedical Sciences and "U.A. IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805 Madrid, Spain.
The use of nucleoside 2'-deoxyribosyltransferases (NDTs) as biocatalysts for the industrial synthesis of nucleoside analogues is often hindered by their strict preference for 2'-deoxyribonucleosides. We now show that a highly versatile purine nucleoside 2'-deoxyribosyltransferase from Trypanosoma brucei (TbPDT) can also accept ribonucleosides as substrates, most likely because of the distinct role played by Asn53 at a position that is usually occupied by Asp in other NDTs. Moreover, this unusual activity was improved ~3-fold by introducing a single amino acid replacement at position 5 following a structure-guided approach. Biophysical and biochemical characterization revealed that the TbPDTY5F variant is a homodimer that displays maximum activity at 50 oC and pH 6.5 and shows a remarkably high melting temperature of 69 oC. Substrate specificity studies demonstrated that 6-oxopurine ribonucleosides are the best donors (inosine > guanosine >> adenosine) whereas no significant preferences exist between 6-aminopurines and 6-oxopurines as base acceptors. In contrast, no transferase activity could be detected on xanthine and 7-deaza purines. TbPDTY5F was successfully employed in the synthesis of a wide range of modified ribonucleosides containing different purine analogues.
© 2019 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
PMID: 31264760 10.1002/cbic.201900397
[Supporting Information]
(F)uridylylated peptides linked to VPg1 of foot-and-mouth disease virus (FMDV): design, synthesis and x-ray crystallography of the complexes
with FMDV RNA-dependent RNA polymerase
de Castro S1, Ferrer-Orta C2, Mills A3, Fernández-Cureses G1, Gago F3, Verdaguer N2, Camarasa MJ.1
Author information:
1 Instituto de Química Médica (IQM-CSIC), Juan de la Cierva 3, E-28006 Madrid, Spain. sonia@iqm.csic.es.
2 Institut de Biologia Molecular de Barcelona (IBMB-CSIC), Parc Científic de Barcelona, Josep Samitier 1-5, 08028 Barcelona, Spain.
3 Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain.
Foot-and Mouth disease virus (FMDV) is an RNA virus belonging to the Picornaviridae family that contains three small viral proteins (VPgs), named VPg1, VPg2 and VPg3, linked to the 5'-end of the viral genome. These VPg proteins act as primers for RNA replication, which is initiated by the consecutive binding of two UMP molecules to the hydroxyl group of Tyr3 in VPg. This process, termed uridylylation, is catalyzed by the viral RNA-dependent RNA polymerase named 3Dpol. 5-Fluorouridine triphosphate (FUTP) is a potent competitive inhibitor of VPg uridylylation. Peptide analysis showed FUMP covalently linked to the Tyr3 of VPg. This fluorouridylylation prevents further incorporation of the second UMP residue. The molecular basis of how the incorporated FUMP blocks the incorporation of the second UMP is still unknown. To investigate the mechanism of inhibition of VPg uridylylation by FUMP, we have prepared a simplified 15-mer model of VPg1 containing FUMP and studied its X-ray crystal structure in complex with 3Dpol. Unfortunately, the fluorouridylylated VPg1 was disordered and not visible in the electron density maps; however, the structure of 3Dpol in the presence of VPg1-FUMP showed an 8-Å movement of the β9-α11 loop of the polymerase towards the active site cavity relative to the complex of 3Dpol with VPg1-UMP. The conformational rearrangement of this loop preceding the 3Dpol B motif seems to block the access of the template nucleotide to the catalytic cavity. This result may be useful in the design of new antivirals against not only FMDV but also other picornaviruses, since all members of this family require the uridylylation of their VPg proteins to initiate the viral RNA synthesis.
PMID: 31247979 10.3390/molecules24132360
| |
Structure reported in this article: 62SL | .
Synthesis and cytotoxicity of 7,9-O-linked macrocyclic C-seco taxoids.
Zhao Y1, Wang TE1, Mills A2, Gago F2, Fang WS.1*
Author information:
1 State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College , Beijing 100050, People's Republic of China.
2 Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain.
A series of novel 7,9-O-linked macrocyclic taxoids together with modification at the C2 position were synthesized, and their cytotoxicities against drug-sensitive and P-glycoprotein and βIII-tubulin overexpressed drug-resistant cancer cell lines were evaluated. It is demonstrated that C-seco taxoids conformationally constrained via carbonate containing-linked macrocyclization display increased cytotoxicity on drug-resistant tumors overexpressing both βIII and P-gp, among which compound 22b, bearing a 2-m-methoxybenzoyl group together with a five-atom linker, was identified as the most potent. Molecular modeling suggested the improved cytotoxicity of 22b results from enhanced favorable interactions with the T7 loop region of βIII.
PMID: 31181726 10.3390/molecules24112161
Unravelling the covalent binding of zampanolide and taccalonolide AJ to a minimalist representation of a human microtubule.
Sánchez-Murcia PA1,2, Mills A1, Cortés-Cabrera Á1, Gago F.1
Author information:
1 Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain.
2 Institute of Theoretical Chemistry, University of Vienna, Währinger Str. 17, 1090, Vienna, Austria.
Many natural products target mammalian tubulin but only a few can form a covalent bond and hence irreversibly affect microtubule function. Among them, zampanolide (ZMP) and taccalonolide AJ (TAJ) stand out, not only because they are very potent antitumor agents but also because the adducts they form with β-tubulin have been structurally characterized in atomic detail. By applying model building techniques, molecular orbital calculations, molecular dynamics simulations and hybrid QM/MM methods, we have gained insight into the 1,2- and 1,4-addition reactions of His229 and Asp226 to ZMP and TAJ, respectively, in the taxane-binding site of β-tubulin. The experimentally inaccessible precovalent complexes strongly suggest a water-mediated proton shuttle mechanism for ZMP adduct formation and a direct nucleophilic attack by the carboxylate of Asp226 on C22 of the C22R,C23R epoxide in TAJ. The M-loop, which is crucially important for interprotofilament interactions, is structured into a short helix in both types of complexes, mostly as a consequence of the fixation of the phenol ring of Tyr283 and the guanidinium of Arg284. As a side benefit, we obtained evidence supporting the existence of a commonly neglected intramolecular disulfide bond between Cys241 and Cys356 in β-tubulin that contributes to protein compactness and is absent in the βIII isotype associated with resistance to taxanes and other drugs.
PMID: 31152293 10.1007/s10822-019-00208-w
[Supplementary Material]
Viral engagement with host (co-)receptors blocked by a novel class of tryptophan dendrimers that targets the 5-fold-axis of the enterovirus-A71 capsid.
Sun L1, Lee H2, Thibaut HJ1, Lanko K1, Rivero-Buceta E3, Bator C2, Martínez-Gualda B3, Dallmeier K1, Delang L1, Leyssen P1, Gago F4, San-Félix A3, Hafenstein S2,5, Mirabelli C1, Neyts J.1
Author information:
1 KU Leuven-University of Leuven, Department of Microbiology and Immunology, Rega Institute for Medical Research, Laboratory of Virology and Chemotherapy, Leuven, Belgium.
2 Department of Biochemistry and Molecular Biology, Huck Institutes of the Life Sciences, The Pennsylvania State University, University Park, Pennsylvania, United States of America.
3 Instituto de Química Médica, Consejo Superior de Investigaciones Científicas (IQM, CSIC), Madrid, Spain.
4 Department of Biomedical Sciences, School of Medicine and Health Sciences, University of Alcalá, Unidad Asociada IQM-CSIC, Madrid, Spain.
5 Department of Medicine, The Pennsylvania State University College of Medicine, Hershey, Pennsylvania, United States of America
Enterovirus A71 (EV-A71) is a non-polio neurotropic enterovirus with pandemic potential. There are no antiviral agents approved to prevent or treat EV-A71 infections. We here report on the molecular mechanism by which a novel class of tryptophan dendrimers inhibits (at low nanomolar to high picomolar concentration) EV-A71 replication in vitro. A lead compound in the series (MADAL385) prevents binding and internalization of the virus but does not, unlike classical capsid binders, stabilize the particle. By means of resistance selection, reverse genetics and cryo-EM, we map the binding region of MADAL385 to the 5-fold vertex of the viral capsid and demonstrate that a single molecule binds to each vertex. By interacting with this region, MADAL385 prevents the interaction of the virus with its cellular (co-)receptors PSGL1 and heparan sulfate, thereby blocking the attachment of EV-A71 to the host cells.
PMID: 31071193 10.1371/journal.ppat.1007760
[Supporting Information]
| |
Cryo-EM structures reported by this article: 6UH1 (EVA71 strain 11316 capsid); 6DIZ (EV-A71 strain 11316 complexed with tryptophan dendrimer MADAL_0385) | .
Pyrrolopyrimidine vs imidazole-phenyl-thiazole scaffolds in non-peptidic dimerization inhibitors of Leishmania infantum trypanothione reductase.
Revuelto A1, Ruiz-Sanquiteria M1, De Lucio H2, Gamo AM1, Carriles AA3, Gutiérrez K1, Sánchez-Murcia PA4, Hermoso JA3, Gago F4, Camarasa MJ1, Jiménez-Ruiz A2, Velázquez S.1
Author information: 1Instituto de Química Médica (IQM-CSIC , Madrid E-28006, Spain. 2Departamento de Biología de Sistemas, Universidad de Alcalá, Alcalá de
Henares, Madrid E-28805, Spain. 3Department of Crystallography and Structural Biology, Institute of Physical Chemistry "Rocasolano" (IQFR-CSIC), Madrid E-28006, Spain. 4Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, Alcalá de Henares, Madrid E-28805, Spain.
Disruption of protein-protein interactions of essential oligomeric enzymes by small molecules represents a significant challenge. We recently reported some linear and cyclic peptides derived from an α-helical region present in the homodimeric interface of Leishmania infantum trypanothione reductase (Li-TryR) that showed potent effects on both dimerization and redox activity of this essential enzyme. Here we describe our first steps towards the design of non-peptidic small-molecule Li-TryR dimerization disruptors using a proteomimetic approach. The pyrrolopyrimidine and the 5-6-5 imidazole-phenyl-thiazole α-helix-mimetic scaffolds were suitably decorated with substituents that could mimic three key residues (K, Q and I) of the linear peptide prototype (PKIIQSVGIS-Nle-K-Nle). Extensive optimization of previously described synthetic methodologies was required. A library of 15 compounds bearing different hydrophobic alkyl and aromatic substituents was synthesized. The imidazole-phenyl-thiazole-based analogues outperformed the pyrrolopyrimidine-based derivatives in both inhibiting the enzyme and killing extracellular and intracellular parasites in cell culture. The most active imidazole-phenyl-thiazole compounds 3e and 3f inhibit Li-TryR and prevent growth of the parasites at low micromolar concentrations similar to those required by the peptide prototype. The intrinsic fluorescence of these compounds inside the parasites visually demonstrates their good permeability in comparison with previous peptide-based Li-TryR dimerization disruptors.
PMID: 30983322 10.1021/acsinfecdis.8b00355
[Supplementary Information]
| |
Crystal structure reported in this article: 6I7N (Li-TryR:2f complex) | .
Cold-induced aldimine bond cleavage by Tris in Bacillus subtilis alanine racemase.
Bernardo-García N, Sánchez-Murcia PA, Espaillat A, Martínez-Caballero CS, Cava F, Hermoso J, Gago F.
Departamento de Ciencias Biomédicas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. federico.gago@uah.es.
Pyridoxal 5'-phosphate (PLP) is a versatile cofactor involved in a large variety of enzymatic processes. Most of PLP-catalyzed reactions, such as those of alanine racemases (AlaRs), present a common resting state in which the PLP is covalently bound to an active-site lysine to form an internal aldimine. The crystal structure of BsAlaR grown in the presence of Tris lacks this covalent linkage and the PLP cofactor appeared deformylated. However, loss of activity in a Tris buffer only occurred after the solution was frozen prior to carrying out the enzymatic assay. This evidence strongly suggests that Tris can access the active site at subzero temperatures and behave as an alternate racemase substrate leading to mechanism-based enzyme inactivation, a hypothesis that is supported by additional X-ray structures and theoretical results from QM/MM calculations. Taken together, our findings highlight a possibly underappreciated role for a common buffer component widely used in biochemical and biophysical experiments.
PMID: 30977502 10.1039/C9OB00223E
[Supplementary Information]
| |
Crystal structures reported by this article: 5IRP (BsAlaR in Tris) / 6Q70 (BsAlaR in HEPES) / 6Q71 (BsAlaR in Bis-Tris propane) / 6Q72 (BsAlaR in PEG/Mg2+) | .
Identification of NEK3 and MOK as novel targets for lithium.
Bravo A1,2, de Lucio H2, Sánchez-Murcia PA1, Jiménez-Ruiz A2, Petrone PM3, Gago F1, Cortés Cabrera A.1,3
1 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Spain.
2 Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Spain.
3 Pharma Research & Early Development (PRED). Roche Innovation Center Basel, Basel, Switzerland.
Lithium ion, commonly used as the carbonate salt in the treatment of bipolar disorders, has been identified as an inhibitor of several kinases, including Glycogen Synthase Kinase-3β, for almost 20 years. However, both the exact mechanism of enzymatic inhibition and its apparent specificity for certain metalloenzymes are still a matter of debate. A data-driven hypothesis is presented that accounts for the specificity profile of kinase inhibition by lithium in terms of the presence of a unique protein environment in the magnesium binding site. This hypothesis has been validated by the discovery of two novel potential targets for lithium, namely NEK3 and MOK, which are related to neuronal function.
PMID: 30667602 10.1111/cbdd.13487
Binding of eEF1A2 to the RNA-dependent protein kinase PKR modulates its activity and promotes tumour cell survival.
Losada A, Muñoz-Alonso MJ, Martínez-Díez M, Gago F, Domínguez JM, Martínez-Leal JF, Galmarini CM.
Departamento de Biología Celular y Farmacogenómica, Pharma Mar S.A., Colmenar Viejo, 28770 Madrid, Spain; Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, 28805 Madrid, Spain.
BACKGROUND: Through several not-fully-characterised moonlighting functions, translation elongation factor eEF1A2 is known to provide a fitness boost to cancer cells. Furthermore, eEF1A2 has been demonstrated to confer neoplastic characteristics on preneoplastic, nontumourigenic precursor cells. We have previously shown that eEF1A2 is the target of plitidepsin, a marine drug currently in development for cancer treatment. Herein, we characterised a new signalling pathway through which eEF1A2 promotes tumour cell survival.
METHODS: Previously unknown binding partners of eEF1A2 were identified through co-immunoprecipitation, high-performance liquid chromatography-mass spectrometry and proximity ligation assay. Using plitidepsin to release eEF1A2 from those protein complexes, their effects on cancer cell survival were analysed in vitro.
RESULTS: We uncovered that double-stranded RNA-activated protein kinase (PKR) is a novel eEF1A2-interacting partner whose proapoptotic
effect is hindered by the translation factor, most likely through sequestration and inhibition of its kinase activity. Targeting eEF1A2 with plitidepsin releases PKR from the complex, facilitating its activation and triggering a mitogen-activated protein kinase signalling cascade together with a nuclear factor-κB-dependent activation of the extrinsic apoptotic pathway, which lead to tumour cell death.
CONCLUSIONS: Through its binding to PKR, eEF1A2 provides a survival boost to cancer cells, constituting an Achilles heel that can
be exploited in anticancer therapy.
PMID: 30420615 10.1038/s41416-018-0336-y
Enzymatic synthesis of therapeutic nucleosides using a highly versatile purine nucleoside 2'-deoxyribosyltransferase from Trypanosoma brucei
Pérez E, Sánchez-Murcia P, Jordaan J, Blanco MD, Mancheño JM, Gago F, Fernández-Lucas J.
The use of enzymes for the synthesis of nucleoside analogues offers several advantages over multistep chemical methods, including chemo-, regio- and stereoselectivity as well as milder reaction conditions. Herein, the production, characterization and utilization of a purine nucleoside 2' deoxyribosyltransferase (PDT) from Trypanosoma brucei are reported. TbPDT is a dimer which displays not only excellent activity and stability over a broad range of temperatures (50-70 ℃), pH (4-7) and ionic strength (0-500 mM NaCl) but also an unusual high stability under alkaline con-ditions (pH 8-10). TbPDT is shown to be proficient in the biosynthesis of numerous therapeutic nucleosides, including didanosine, vidarabine, cladribine, fludarabine and nelarabine. The structure-guided replacement of Val11 with either Ala or Ser resulted in variants with 2.8-fold greater activity. TbPDT was also covalently immobilized on glutaraldehyde-activated magnetic microspheres. MTbPDT3 was selected as the best derivative (4200 IU/g, activity recovery of 22%), and could be easily recaptured and recycled for >25 reactions with negligible loss of activity. Finally, MTbPDT3 was successfully employed in the expedient synthesis of several nucleoside analogues. Taken together, our results support the notion that TbPDT has good potential as an industrial biocatalyst for the synthesis of a wide range of therapeutic nucleosides through an efficient and environmentally friendly methodology.
PMID: xxx 10.1002/cctc.201800775
Investigation of the complexation between cyclodextrins and medetomidine enantiomers by capillary electrophoresis, NMR spectroscopy and molecular
modeling.
Krait S1, Salgado A2, Chankvetadze B3, Gago F3, Scriba GKE.1
Author information:
1 Friedrich-Schiller-University Jena, Department of Pharmaceutical/Medicinal Chemistry, Philosophenweg 14, 07743 Jena, Germany. 2NMR Spectroscopy Centre (CERMN), CAI Químicas, Faculty of Pharmacy, University of Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. a.salgado@uah.es. 3 Institute of Physical and Analytical Chemistry, School of Exact and Natural Sciences, Tbilisi State University, 0179 Tbilisi, Georgia. 4 Department of Biomedical Sciences (Unidad Asociada IQM-CSIC) and Instituto de Investigación Química "Andrés M. del Río" (IQAR), University of Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
The migration order of the enantiomers of medetomidine in the presence of cyclodextrins studied by capillary electrophoresis in phosphate buffer, pH 2.5, depended on the cavity size and the substitution pattern of the cyclodextrins. Opposite migration order was observed in the presence of β-cyclodextrin (β-CD) and γ-cyclodextrin (γ-CD) as well as randomly sulfated β-CD (S-β-CD) and heptakis(6-O-sulfo)-β-CD (HS-β-CD). This could be rationalized by the fact that dexmedetomidine formed more stable complexes with β-CD and S-β-CD, while levomedetomidine interacted stronger with γ-CD and HS-β-CD. The structure of the complexes was derived from rotating frame nuclear Overhauser (ROESY) experiments for β-CD, γ-CD and HS-β-CD. In the case of the native CDs, the phenyl ring of medetomidine entered the cavity through the wider secondary rim of the CDs, whereas the protonated imidazole ring was positioned inside the CD cavity interacting with the sulfate groups of HS-β-CD. Furthermore, molecular dynamics calculations also suggested opposite affinities of the medetomidine enantiomers toward β-CD and γ-CD.
PMID: 30055912 10.1016/j.chroma.2018.06.010
Characterization of an atypical, thermostable, organic solvent- and acid-tolerant 2'-deoxyribosyltransferase from Chroococcidiopsis thermalis.
Del Arco J1, Sánchez-Murcia PA2, Mancheño JM3, Gago F4, Fernández-Lucas J.5,6
Author information:
1 Applied Biotechnology Group, Universidad Europea de Madrid, Urbanización El Bosque, c/ Tajo, s/n, Villaviciosa de Odón, 28670 Madrid, Spain.
2 Institute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, 1090 Vienna, Austria.
3 Department of Crystallography and Structural Biology, Rocasolano Institute (CSIC), c/ Serrano 119, 28006 Madrid, Spain.
4 Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805 Madrid, Spain.
5 Applied Biotechnology Group, Universidad Europea de Madrid, Urbanización El Bosque, c/ Tajo, s/n, Villaviciosa de Odón, 28670 Madrid, Spain. jesus.fernandez2@universidadeuropea.es
6 Grupo de Investigación en Desarrollo Agroindustrial Sostenible, Universidad de la Costa, CUC, Calle 58 # 55-66, Barranquilla, 080002 Colombia. jesus.fernandez2@universidadeuropea.es.
In our search for thermophilic and acid-tolerant nucleoside 2'-deoxyribosyltransferases (NDTs), we found a good candidate in an enzyme encoded by Chroococcidiopsis thermalis PCC 7203 (CtNDT). Biophysical and biochemical characterization revealed CtNDT as a homotetramer endowed with good activity and stability at both high temperatures (50-100 ℃) and a wide range of pH values (from 3 to 7). CtNDT recognizes purine bases and their corresponding 2'-deoxynucleosides but is also proficient using cytosine and 2'-deoxycytidine as substrates. These unusual features preclude the strict classification of CtNDT as either a type I or a type II NDT and further suggest that this simple subdivision may need to be updated in the future. Our findings also hint at a possible link between oligomeric state and NDT's substrate specificity. Interestingly from a practical perspective, CtNDT displays high activity (80-100%) in the presence of several water-miscible co-solvents in a proportion of up to 20% and was successfully employed in the enzymatic production of several therapeutic nucleosides such as didanosine, vidarabine, and cytarabine.
PMID: 29872887 10.1007/s00253-018-9134-y
[Supplementary content]
High-affinity ligands of the colchicine domain in tubulin based on a structure-guided design
Bueno O, Estévez Gallego J, Martins S, Prota AE, Gago F, Gómez-SanJuan A, Camarasa MJ, Barasoain I, Steinmetz MO, Díaz JF, Pérez-Pérez MJ, Liekens S, Priego EM.
Microtubule-targeting agents that bind at the colchicine-site of tubulin are of particular interest in antitumoral therapy due to their dual mechanism of action as antimitotics and vascular disrupting agents. Cyclohexanediones derivatives have been described as a new family of colchicine-domain binders with an association constant to tubulin similar to that of colchicine. Here, the high-resolution structures of tubulin in complex with cyclohexanediones TUB015 and TUB075 were solved by X-ray crystallography. A detailed analysis of the tubulin-TUB075 interaction by means of computational affinity maps allowed the identification of two additional regions at the binding site that were addressed with the design and synthesis of a new series of cyclohexanediones with a distal 2-substituted benzofurane. These new compounds showed potent antiproliferative activity with IC50 values in the nM range, arrested cell cycle progression at the G2/M phase and induced apoptosis at sub µM concentrations. Moreover, they caused the destruction of a preformed vascular network in vitro and inhibited the migration of endothelial cells at non-toxic concentrations. Finally, these compounds displayed high affinity for tubulin as substantiated by a Kb value of 2.87 × 108 M-1 which, to the best of our knowledge, represents the highest binding constant measured to date for a colchicine-domain ligand.
PMID:29523799 10.1038/s41598-018-22382-x
| |
Structures reported by this article: 6FKJ (T2R-TTL-TUB075) / 6FKL (T2R-TTL-TUB015) | .
Trypanothione reductase inhibition and anti-leishmanial activity of all-hydrocarbon stapled α-helical peptides with improved proteolytic stability
Ruiz-Santaquiteria M1, de Castro S1, Toro MA2, de Lucio H2, Gutiérrez KJ2, Sánchez-Murcia PA3, Jiménez, MA.4, Gago F3, Jiménez-Ruiz A2, Camarasa MJ1, Velázquez S.1
Author information:
1 Instituto de Química Médica (IQM-CSIC), E-28006 Madrid, Spain. 2 Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 3 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 4 Instituto de Química Física Rocasolano, Consejo Superior de Investigaciones Científicas (CSIC), E-28006 Madrid, Spain. Electronic address: iqmsv29@iqm.csic.es.
Trypanothione reductase (TryR) is a well-established target in the search for novel antitrypanosomal and antileishmanial agents. We have previously identified linear and lactam-bridged 13-residue peptides derived from an ?-helical region making up part of the dimeric interface of Leishmania infantum TryR (Li-TryR) which prevent trypanothione reduction by disrupting enzyme dimerization. We now show that i,i + 4 side-chain cross-linking with an all-hydrocarbon staple stabilizes the helical structure of these peptides and significantly improves their resistance to protease cleavage relative to previous linear and cyclic lactam analogues. Interestingly, replacement of the amide bridge by the hydrocarbon staple at the same cyclization positions generates derivatives (2 and 3) that similarly inhibit oxidoreductase activity of the enzyme but unexpectedly stabilize the TryR homodimer. The most proteolytically stable peptide 2 covalently linked to oligoarginines displayed potent in vitro leishmanicidal activity against L. infantum parasites.
PMID: 29501944 10.1016/j.ejmech.2018.02.071
[Supplementary data]
Engineering Erg10 thiolase from Saccharomyces cerevisiae as a synthetic toolkit for the production of branched-chain alcohols.
Torres-Salas P, Bernal V, López-Gallego F, Martínez-Crespo J, Sánchez-Murcia PA, Barrera V, Morales-Jiménez R, García-Sánchez A, Mañas-Fernández A, Seoane JM, Sagrera Polo M, Miranda JD, Calvo J, Huertas S, Torres JL, Alcalde-Bascones A, González-Barrera S, Gago F, Morreale A, González-Barroso MdM.
Thiolases catalyze the condensation of acyl-CoA thioesters through the Claisen condensation reaction. The best described enzymes usually yield linear condensation products. Using a combined computational/experimental approach, and guided by structural information, we have studied the potential of thiolases to synthesize branched compounds. We have identified a bulky residue located at the active site that blocks proper accommodation of substrates longer than acetyl-CoA. Amino acid replacements at such position exert effects on the activity and product selectivity of the enzymes that are highly dependent on protein scaffold. Among the set of five thiolases studied, Erg10 thiolase from Saccharomyces cerevisiae showed no acetyl-CoA/butyryl-CoA branched condensation activity, but variants at position F293 resulted in the most active and selective biocatalysts for this reaction. This is the first time that a thiolase has been engineered to synthesize branched compounds. These novel enzymes enrich the toolbox of combinatorial (bio)chemistry, paving the way for manufacturing a variety of α-substituted synthons. As a proof of concept, we have engineered Clostridium's 1-butanol pathway to obtain 2-ethyl-1-butanol, an alcohol which is interesting as a branched model compound.
PMID: 29360348 10.1021/acs.biochem.7b01186
[Supplementary files]
A series of enthalpically optimized docetaxel analogues exhibiting enhanced antitumor activity and water solubility.
Ma YT1, Yang Y2, Cai P1, Sun DY1, Sánchez-Murcia PA3, Zhang XY1, Jia WQ1, Lei L2, Guo M2, Gago F3, Wang H2, Fang WS1.
Author information:
1State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences and Peking Union Medical College , Beijing 100050, People's Republic of China.
2Key Laboratory of Molecular Pharmacology and Drug Evaluation, Yantai University, Ministry of Education, Collaborative Innovation Center of Advanced Drug Delivery System and Biotech Drugs in Universities of Shandong, Yantai University , Yantai 264005, People's Republic of China.
3Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al Instituto de Química Médica del CSIC, Universidad de Alcalá , E-28805 Alcalá de Henares, Madrid, Spain.
A dual-purpose strategy aimed at enhancing the binding affinity for microtubules and improving the water solubility of docetaxel led to the design and synthesis of a series of C-2- and C-3'-modified analogues. Both aims were realized when the C-3' phenyl group present in docetaxel was replaced with a propargyl alcohol. The resulting compound, 3f, was able to overcome drug resistance in cultured P-gp-overexpressing tumor cells and showed greater activity than docetaxel against drug-resistant A2780/AD ovarian cancer xenografts in mice. In addition, the considerably lower hydrophobicity of 3f relative to both docetaxel and paclitaxel led to better aqueous solubility. A molecular model of tubulin-bound 3f revealed novel hydrogen-bonding interactions between the propargyl alcohol and the polar environment provided by the side chains of Ser236, Glu27, and Arg320.
PMID: 29359935 10.1021/acs.jnatprod.7b00857
[Supporting Information]
Structural rationale for the chiral separation and migration order reversal of clenpenterol enantiomers in capillary electrophoresis using two different β-cyclodextrins
Salgado A1, Tatunashvili E2, Gogolashvili A2, Chankvetadze B2, Gago F3.
Author information:
1 NMR Spectroscopy Centre (CERMN), CAI Químicas, Faculty of Pharmacy, University of Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. a.salgado@uah.es. 2 Institute of Physical and Analytical Chemistry, School of Exact and Natural Sciences, Tbilisi State University, 0179 Tbilisi, Georgia. 3 Department of Biomedical Sciences (Unidad Asociada IQM-CSIC) and Instituto de Investigación Química "Andrés M. del Río" (IQAR), University of Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
NMR spectroscopy experiments, molecular dynamics simulations, and theoretical chemistry calculations provide insight into the structural and energetic determinants of the distinct binding of clenpenterol enantiomers to two cyclodextrins and the migration order reversal of their respective inclusion complexes in capillary electrophoresis.
PMID: 29022621 10.1039/c7cp04761d
[Supplementary files]
Improved proteolytic stability and potent activity against Leishmania infantum trypanothione reductase of α/β-peptide foldamers conjugated to cell-penetrating peptides
de Lucio H1, Gamo, AM2, Ruiz-Santaquiteria M2, de Castro S2, Sánchez-Murcia PA3, Toro MA1, Gutiérrez KJ1, Gago F3, Jiménez-Ruiz A1, Camarasa MJ2, Velázquez S4.
Author information:
1 Instituto de Química Médica (IQM-CSIC), E-28006 Madrid, Spain. 2 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 3 Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 4 Instituto de Química Médica (IQM-CSIC), E-28006 Madrid, Spain. Electronic address: iqmsv29@iqm.csic.es.
The objective of the current study was to enhance the proteolytic stability of peptide-based inhibitors that target critical protein-protein interactions at the dimerization interface of Leishmania infantum trypanothione reductase (Li-TryR) using a backbone modification strategy. To achieve this goal we carried out the synthesis, proteolytic stability studies and biological evaluation of a small library of α/β3-peptide foldamers of different length (from 9-mers to 13-mers) and different α→β substitution patterns related to prototype linear α-peptides. We show that several 13-residue α/β3-peptide foldamers retain inhibitory potency against the enzyme (in both activity and dimerization assays) while they are far less susceptible to proteolytic degradation than an analogous α-peptide. The strong dependence of the binding affinities for Li-TryR on the length of the α/β-peptides is supported by theoretical calculations on conformational ensembles of the resulting complexes. The conjugation of the most proteolytically stable α/β-peptide with oligoarginines results in a molecule with potent activity against L. infantum promastigotes and amastigotes.
PMID: 29017116 10.1016/j.ejmech.2017.09.032
[Supplementary content]
Structure-based domain assignment in Leishmania infantum EndoG: characterization of a pH-dependent regulatory switch and a C-terminal extension that largely dictates DNA substrate preferences.
Oliva C1, Sánchez-Murcia PA2, Rico E1, Bravo A2, Menéndez M3, Gago F2, Jiménez-Ruiz A1.
Author information:
1 Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
2 Departamento de Ciencias Biomédicas y "Unidad Asociada IQM-CSIC", Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain.
3 Instituto de Química Física Rocasolano, Consejo Superior de Investigaciones Científicas (CSIC), E-28006 Madrid, Spain.
Mitochondrial endonuclease G from Leishmania infantum (LiEndoG) participates in the degradation of double-stranded DNA (dsDNA) during parasite cell death and is catalytically inactive at a pH of 8.0 or above. The presence, in the primary sequence, of an acidic amino acid-rich insertion exclusive to trypanosomatids and its spatial position in a homology-built model of LiEndoG led us to postulate that this peptide stretch might act as a pH sensor for self-inhibition. We found that a LiEndoG variant lacking residues 145-180 is indeed far more active than its wild-type counterpart at pH values >7.0. In addition, we discovered that (i) LiEndoG exists as a homodimer, (ii) replacement of Ser211 in the active-site SRGH motif with the canonical aspartate from the DRGH motif of other nucleases leads to a catalytically deficient enzyme, (iii) the activity of the S211D variant can be restored upon the concomitant replacement of Ala247 with Arg and (iv) a C-terminal extension is responsible for the observed preferential cleavage of single-stranded DNA (ssDNA) and ssDNA-dsDNA junctions. Taken together, our results support the view that LiEndoG is a multidomain molecular machine whose nuclease activity can be subtly modulated or even abrogated through architectural changes brought about by environmental conditions and interaction with other binding partners.
PMID: 28911117  PMCID: PMC5587815 10.1093/nar/gkx629
PMCID: PMC5587815 10.1093/nar/gkx629
Structural rationale for the cross-resistance of tumor cells bearing the A399V variant of elongation factor eEF1A1 to the structurally unrelated didemnin B, ternatin, nannocystin A and ansatrienin B.
Sánchez-Murcia PA1,2, Cortés-Cabrera Á1,3, Gago F4
Author information:
1 Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain.
2 Present address: Institute of Theoretical Chemistry, University of Vienna, Währinger Str. 17, 1090, Vienna, Austria.
3 Present addressComputational Chemistry-UK, RD Platform Technology & Science, GSK Medicines Research Centre, Gunnels Wood Road, Stevenage, Hertfordshire, SG1 2NY, UK.
4Area of Pharmacology, Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, 28805, Madrid, Spain. federico.gago@uah.es.
At least four classes of structurally distinct natural products with potent antiproliferative activities target the translation elongation factor eEF1A1, which is best known as the G-protein that delivers amino acyl transfer RNAs (aa-tRNAs) to ribosomes during mRNA translation. We present molecular models in atomic detail that provide a common structural basis for the high-affinity binding of didemnin B, ternatin, ansatrienin B and nannocystin A to eEF1A1, as well as a rationale based on molecular dynamics results that accounts for the deleterious effect of replacing alanine 399 with valine. The proposed binding site, at the interface between domains I and III, is eminently hydrophobic and exists only in the GTP-bound conformation. Drug binding at this site is expected to disrupt neither loading of aa-tRNAs nor GTP hydrolysis but would give rise to stabilization of this particular conformational state, in consonance with reported experimental findings. The experimental solution of the three-dimensional structure of mammalian eEF1A1 has proved elusive so far and the highly homologous eEF1A2 from rabbit muscle has been crystallized and solved only as a homodimer in a GDP-bound conformation. Interestingly, in this dimeric structure the large interdomain cavity where the drugs studied are proposed to
bind is occupied by a mostly hydrophobic α-helix from domain I of the same monomer. Since binding of this α-helix and any of these drugs to domain III of eEF1A(1/2) is, therefore, mutually exclusive and involves two distinct protein conformations, one intriguing possibility that emerges from our study is that the potent antiproliferative effect of these natural products may arise not only from inhibition of protein synthesis, which is the current dogma, but also from interference with some other non-canonical functions. From this standpoint, this type of drugs could be considered antagonists of eEF1A1/2 oligomerization, a hypothesis that opens up novel areas of research.
PMID: 28900796 10.1007/s10822-017-0066-x
Making sense of the past: hyperstability of ancestral thioredoxins explained by free energy simulations.
Cortés Cabrera A, Sánchez-Murcia PA, Gago F.
Author information:
Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, E-28871 Alcalá de Henares, Spain.
Thioredoxin (Trx), a small and globular protein, is present in all kinds of organisms, from Archea to higher mammals. Throughout evolution, the Trx sequence has undergone subtle modifications to adapt to varying environmental conditions. The high degree of sequence conservation makes Trx very amenable to ancestral protein reconstruction techniques. In this work, we address the study of the structural and energetic determinants of thermostability in E. coli Trx using a dataset of mutations inspired by ancestral reconstruction. We compute, from first principles, the expected contribution of 19 different amino acid substitutions to the stability (ΔΔG) and the melting temperature (ΔTm) of the protein. We also describe the specific changes in structure and protein dynamics responsible for the stabilizing or destabilizing effects of these mutations. Our results point to local and independent changes for most of the variants. Our predictions are accurate enough to substantiate the proposal of new hypotheses regarding evolutionary relationships between mutations, as in the case of T89R, P68A and G74S or K90L and F102A, and reach beyond the initial set to suggest improved variants, such as K90I or K90Y.
PMID: 28825743 10.1039/C7CP03659K
[Supplementary files]
2'-Deoxyribosyltransferase from Leishmania mexicana, an efficient biocatalyst for one-pot, one-step synthesis of nucleosides from poorly soluble purine bases.
Crespo N1,2, Sánchez-Murcia PA3,4, Gago F4, Cejudo-Sanches J2, Galmes MA2, Fernández-Lucas J5,6, Mancheño JM7.
Author information:
1 Department of Crystallography and Structural Biology, Institute Rocasolano (CSIC), Serrano 119, E-28006 Madrid, Spain.
2 Applied Biotechnology Group, European University of Madrid, E-28670 Villaviciosa de Odón, Madrid, Spain.
3 Institute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, 1090 Vienna, Austria.
4 Department of Biomedical Sciences and "Unidad Asociada IQM-CSIC", School of Medicine and Health Sciences, University of Alcalá, E-28871 Alcalá de Henares, Spain.
5 Applied Biotechnology Group, European University of Madrid, E-28670 Villaviciosa de Odón, Madrid, Spain. jesus.fernandez2@universidadeuropea.es.
6 Grupo de Investigación en Desarrollo Agroindustrial Sostenible, Department of Agroindustrial Engineering, School of Environmental Sciences, Universidad de la Costa, Cra. 55 #58-66, Barranquilla, Colombia. jesus.fernandez2@universidadeuropea.es.
7 Department of Crystallography and Structural Biology, Institute Rocasolano (CSIC), Serrano 119, E-28006 Madrid, Spain. xjosemi@iqfr.csic.es.
Processes catalyzed by enzymes offer numerous advantages over chemical methods although in many occasions the stability of the biocatalysts becomes a serious concern. Traditionally, synthesis of nucleosides using poorly water-soluble purine bases, such as guanine, xanthine, or hypoxanthine, requires alkaline pH and/or high temperatures in order to solubilize the substrate. In this work, we demonstrate that the 2'-deoxyribosyltransferase from Leishmania mexicana (LmPDT) exhibits an unusually high activity and stability under alkaline conditions (pH 8-10) across a broad range of temperatures (30-70 ℃) and ionic strengths (0-500 mM NaCl). Conversely, analysis of the crystal structure of LmPDT together with comparisons with hexameric, bacterial homologues revealed the importance of the relationships between the oligomeric state and the active site architecture within this family of enzymes. Moreover, molecular dynamics and docking approaches provided structural insights into the substrate-binding mode. Biochemical characterization of LmPDT identifies the enzyme as a type I NDT (PDT), exhibiting excellent activity, with specific activity values 100- and 4000-fold higher than the ones reported for other PDTs. Interestingly, LmPDT remained stable during 36 h at different pH values at 40 ℃. In order to explore the potential of LmPDT as an industrial biocatalyst, enzymatic production of several natural and non-natural therapeutic nucleosides, such as vidarabine (araA), didanosine (ddI), ddG, or 2'-fluoro-2'-deoxyguanosine, was carried out using poorly water-soluble purines. Noteworthy, this is the first time that the enzymatic synthesis of 2'-fluoro-2'-deoxyguanosine, araG, and araH by a 2'-deoxyribosyltransferase is reported.
PMID: 28785897 10.1007/s00253-017-8450-y
[Supplementary content]
| |
Structure reported by this article: 6QAI |
First example of peptides targeting the dimer interface of Leishmania infantum trypanothione reductase with potent in vitro antileishmanial activity
Ruiz-Santaquiteria M1, Sánchez-Murcia PA2, Toro MA3, de Lucio H3, Gutiérrez KJ3, de Castro S1, Carneiro FAC1, Gago F2, Jiménez-Ruiz A3, Camarasa MJ1, Velázquez S4.
Author information:
1 Instituto de Química Médica (IQM-CSIC), E-28006 Madrid, Spain. 2 Área de Farmacología, Departamento de Ciencias Biomédicas, Unidad Asociada al IQM-CSIC, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 3 Departamento de Biología de Sistemas, Universidad de Alcalá, E-28805 Alcalá de Henares, Madrid, Spain. 4 Instituto de Química Médica (IQM-CSIC), E-28006 Madrid, Spain. Electronic address: iqmsv29@iqm.csic.es.
A series of 9-mer and 13-mer amide-bridged cyclic peptides derived from the linear prototype Ac-PKIIQSVGIS-Nle-K-Nle-NH2 (Toro et al. ChemBioChem 2013) has been designed and synthesized by introduction of the lactam between amino acid side chains that are separated by one helical turn (i, i+4). All of these compounds were tested in vitro as both dimerization and enzyme inhibitors of Leishmania infantum trypanothione reductase (Li-TryR). Three of the 13-mer cyclic peptide derivatives (3, 4 and 6) inhibited the oxidoreductase activity of Li-TryR in the low micromolar range and they also disrupted enzyme dimerization. Cyclic analogues 3 and 4 were more resistant to proteases than was the linear prototype. Furthermore, the most potent TryR inhibitors in the linear and cyclic series displayed potent in vitro activity against Leishmania infantum upon conjugation with cationic cell-penetrating peptides. To date, these conjugated peptides can be considered the first example of TryR dimerization inhibitors that are active in cell culture.
PMID: 28431354 10.1016/j.ejmech.2017.04.020
[Supplementary content]
Structural determinants of the dictyostatin chemotype for tubulin binding affinity and antitumor activity against taxane- and epothilone-resistant cancer cells
Trigili C, Barasoain I, Sánchez-Murcia PA, Bargsten K, Redondo-Horcajo M, Nogales A, Gardner NM, Meyer A, Naylor GJ, Gómez-Rubio E, Gago F, Steinmetz MO, Paterson I, Prota AE, Díaz JF.
A combined biochemical, structural, and cell biology characterization of dictyostatin is described, which enables an improved understanding of the structural determinants responsible for the high-affinity binding of this anticancer agent to the taxane site in microtubules (MTs). The study reveals that this macrolide is highly optimized for MT binding and that only a few of the structural modifications featured in a library of synthetic analogues resulted in small gains in binding affinity. The high efficiency of the dictyostatin chemotype in overcoming various kinds of clinically relevant resistance mechanisms highlights its potential for therapeutic development for the treatment of drug-resistant tumors. A structural explanation is advanced to account for the synergy observed between dictyostatin and taxanes on the basis of their differential effects on the MT lattice. The X-ray crystal structure of a tubulin-dictyostatin complex and additional molecular modeling have allowed the rationalization of the structure-activity relationships for a set of synthetic dictyostatin analogues, including the highly active hybrid 12 with discodermolide. Altogether, the work reported here is anticipated to facilitate the improved design and synthesis of more efficacious dictyostatin analogues and hybrids with other MT-stabilizing agents.
PMID: 30023505 10.1021/acsomega.6b00317
| |
Structure reported by this article: 5MF4 |
Modular Architecture and Unique Teichoic Acid Recognition Features of Choline-Binding Protein L (CbpL) Contributing to Pneumococcal Pathogenesis
Gutiérrez-Fernández J, Saleh M, Alcorlo M, Gómez-Mejía A, Pantoja-Uceda D, Treviño MA, Voß F, Abdullah MR, Galán-Bartual S, Seinen J, Sánchez-Murcia PA, Gago F, Bruix M, Hammerschmidt S, Hermoso JA.
The human pathogen Streptococcus pneumoniae is decorated with a special class of surface-proteins known as choline-binding proteins (CBPs) attached to phosphorylcholine (PCho) moieties from cell-wall teichoic acids. By a combination of X-ray crystallography, NMR, molecular dynamics techniques and in vivo virulence and phagocytosis studies, we provide structural information of choline-binding protein L (CbpL) and demonstrate its impact on pneumococcal pathogenesis and immune evasion. CbpL is a very elongated three-module protein composed of (i) an Excalibur Ca2+-binding domain -reported in this work for the very first time-, (ii) an unprecedented anchorage module showing alternate disposition of canonical and non-canonical choline-binding sites that allows vine-like binding of fully-PCho-substituted teichoic acids (with two choline moieties per unit), and (iii) a Ltp_Lipoprotein domain. Our structural and infection assays indicate an important role of the whole multimodular protein allowing both to locate CbpL at specific places on the cell wall and to interact with host components in order to facilitate pneumococcal lung infection and transmigration from nasopharynx to the lungs and blood. CbpL implication in both resistance against killing by phagocytes and pneumococcal pathogenesis further postulate this surface-protein as relevant among the pathogenic arsenal of the pneumococcus.
PMID: 27917891 10.1038/srep38094
|
 |
Foto finalista Concurso Imagin'Action 2016 SBE:
CLIMBING TEICHOIC ACIDS. A vine-like molecular model of some teichoic acid units (green sticks) bound to several copies of Streptococcus pneumoniae choline-binding protein L (dark and light brown surfaces). The complex was characterized by combining X-ray crystallography data (PDB id. 4CNL), symmetry operations in the unit cell and molecular modelling techniques. The red "berries" stand for the choline trimethylammonium nitrogens, which are differentially recognized by means of cation-pi interactions. The figure was generated with PyMOL and overlaid on a watercolor-transformed photograph of a real tree-climbing ivy. |
| |
Structure reported by this article: 4CNL |
Translation Elongation Factor eEF1A2 is a Novel Anticancer Target for the Marine Natural Product Plitidepsin
Losada A, Muñoz-Alonso MJ, García C, Sánchez-Murcia PA, Martínez-Leal JF, Domínguez JM, Lillo MP, Gago F, Galmarini C.
eEF1A2 is one of the isoforms of the α subunit of the eukaryotic Elongation Factor 1. It is overexpressed in human tumors and is endowed with oncogenic properties, favoring tumor cell proliferation while inhibiting apoptosis. We demonstrate that plitidepsin, an antitumor agent of marine origin that has successfully completed a phase-III clinical trial for multiple myeloma, exerts its antitumor activity by targeting eEF1A2. The drug interacts with eEF1A2 with a KD of 80 nM and a target residence time of circa 9 min. This protein was also identified as capable of binding [14C]-plitidepsin in a cell lysate from K-562 tumor cells. A molecular modelling approach was used to identify a favorable binding site for plitidepsin at the interface between domains 1 and 2 of eEF1A2 in the GTP conformation. Three tumor cell lines selected for at least 100-fold more resistance to plitidepsin than their respective parental cells showed reduced levels of eEF1A2 protein. Ectopic expression of eEF1A2 in resistant cells restored the sensitivity to plitidepsin. FLIM-phasor FRET experiments demonstrated that plitidepsin localizes in tumor cells sufficiently close to eEF1A2 as to suggest the formation of drug-protein complexes in living cells. Altogether, our results strongly suggest that eEF1A2 is the primary target of plitidepsin.
PMID: 27713531 10.1038/srep35100
Stepwise simulation of 3,5-dihydro-5-methylidene-4H-imidazol-4-one (MIO) biogenesis in histidine ammonia-lyase.
Sánchez-Murcia PA, Bueren-Calabuig JA, Camacho-Artacho M, Cortés-Cabrera Á, Gago F.
A 3,5-dihydro-5-methylidene-4H-imidazol-4-one (MIO) electrophilic moiety is post-translationally and autocatalytically generated in homotetrameric histidine ammonia-lyase (HAL) and other enzymes containing the tripeptide Ala-Ser-Gly in a suitably positioned loop. The backbone cyclization step is identical to that taking place during fluorophore formation in green fluorescent protein from the tripeptide Ser-Tyr-Gly but dehydration, rather than dehydrogenation by molecular oxygen, is the reaction that gives rise to the mature MIO ring system. To gain additional knowledge about this unique process and shed light on some still unresolved issues we have made use of extensive molecular dynamics simulations and hybrid quantum mechanics/molecular mechanics calculations implementing the self-consistent charge density functional tight-binding method on a fully solvated tetramer of Pseudomonas putida HAL. Our results strongly support that mechanical compression of the reacting loop by neighboring protein residues in the precursor state is absolutely required to prevent formation of inhibitory main-chain hydrogen bonds and to enforce proper alignment of donor and acceptor orbitals for bond creation. The consideration of the protein environment in our computations shows that water molecules, which have been mostly neglected in previous theoretical work, play a highly relevant role in the reaction mechanism, and more importantly, that backbone cyclization resulting from the nucleophilic attack of the Gly amide lone pair to the π* orbital of the Ala carbonyl precedes side-chain dehydration of the central serine.
PMID: 27682658 10.1021/acs.biochem.6b00744
Pro-death activity of a BH3 domain in an aquaporin from the protozoan parasite Leishmania.
Genes CM1, de Lucio H1, Sánchez-Murcia PA2, Gago F2, Jiménez-Ruiz A1.
Author information:
1Departamento de Biología de Sistemas, Universidad de Alcalá, Alcalá de Henares, 28805 Spain; 2 Departamento de Ciencias Biomédicas, Universidad de Alcalá, Alcalá de Henares, 28805 Spain.
News and Commentary
PMID: 27468694 10.1038/cddis.2016.229
A functional BH3 domain in an aquaporin from Leishmania infantum.
Genes CM1, de Lucio H1, González VM2, Sánchez-Murcia PA3, Rico E1, Gago F3, Fasel N4, Jiménez-Ruiz A1.
Author information:
1Departamento de Biología de Sistemas, Universidad de Alcalá, Alcalá de Henares 28805, Spain; 2Laboratory of aptamers, Departamento de Bioquímica-Investigación, IRYCIS-Hospital Ramón y Cajal, Madrid, Spain; 3 Departamento de Ciencias Biomédicas, Universidad de Alcalá, Alcalá de Henares 28805, Spain; 4Department of Biochemistry, University of Lausanne, 155 Chemin des Boveresses, Epalinges 1066, Switzerland.
Despite the absence of sequences showing significant similarity to any of the members of the Bcl-2 family of proteins in protozoa, experiments carried out in yeast or trypanosomatids have demonstrated that ectopic expression of some of these members alters their response to different death stimuli. Because the BH3 domain is the smallest common signature in all the proteins of this family of apoptosis regulators and also because they are essential for molecular interactions between antagonistic members, we looked for sequences with significant similarity to the BH3 motif in the Leishmania infantum genome. Among the top scoring ones, we found the MYLALQNLGDEV amino-acid stretch at the C terminus of a previously described aquaporin, now renamed as Li-BH3AQP. This motif is highly conserved in homologous proteins from other species of the Leishmania genus. The association of Li-BH3AQP with human Bcl-XL was demonstrated by both co-immunoprecipitation and yeast two-hybrid experiments. Ectopic expression of Li-BH3AQP reduced viability of HeLa cells and this deleterious effect was abrogated by the simultaneous overexpression of Bcl-XL. Although we were not able to demonstrate a reduction in parasite viability when the protein was overexpressed in Leishmania promastigotes, a prodeath effect could be observed when the parasites overexpressing Li-BH3AQP were treated with staurosporine or antimycin A. Surprisingly, these parasites were more resistant, compared with wild-type parasites, to hypotonic stress or nutrient deprivation. The prodeath activity was abolished upon replacement of two highly conserved amino acids in this BH3 domain. Taken together, these results point to Li-BH3AQP as the first non-enzymatic protein ever described in trypanosomatids that is involved in cell death.
PMID: 27551533 10.1038/cddiscovery.2016.43
Structural Bioinformatics in Broad-Spectrum Racemases: A New Path in Antimicrobial Research.
Bernardo-García N1, Sánchez-Murcia PA2, Gago F2, Cava F3, Hermoso JA1.
Author information:
1Department of Crystallography and Structural Biology, Instituto de Química Física "Rocasolano", CSIC, c/ Serrano 119, E-28006 Madrid, Spain; 2 Departamento de Ciencias Biomédicas, Universidad de Alcalá, Alcalá de Henares 28805, Spain; 3Department of Molecular Biology and Laboratory for Molecular Infection Medicine Sweden, Umeå Centre for Microbial Research, Umeå University, Umeå, Sweden.
D-amino acids are essential components of the bacterial cell wall and play notable roles in microbiology as regulators, for example in sporulation, biofilm formation or interspecies communication. Racemases are the specific enzymes catalyzing the interconversion of L-amino acids to D-amino acids. While most of racemases are mono-specific, a family of broad-spectrum racemases that can racemize ten of the 19 natural chiral amino acids has been recently reported. These enzymes can interconvert radically different residues such as aliphatic and positively charged residues producing non-canonical D-amino acids. Crystal structures together with bioinformatics allowed identification of the residues defining the molecular footprint in broad-spectrum racemases, the specific features of their active sites and the structural basis of their promiscuity. Here we review the recent knowledge on this family compared with that of the well established alanine racemases. This structural information is a prerequisite for the development of novel drugs against important human pathogens in which broad-spectrum racemases play a key role.
10.2174/1385272819666150810213115
Molecular Interactions and Implications of Aldose Reductase Inhibition by PGA1 and Clinically Used Prostaglandins.
Díez-Dacal B1, Sánchez-Gómez FJ1, Sánchez-Murcia PA2, Milackova I3, Zimmerman T1, Ballekova J3, García-Martín E4, Agúndez JA4, Gharbi S5, Gago F6, Stefek M3, Pérez-Sala D7.
Author information:
1Centro de Investigaciones Biológicas, C.S.I.C.; 2Department of Biomedical Sciences, Universidad de Alcalá; 3Slovak Academy of Sciences; 4Department of Pharmacology, University of Extremadura, Spain; 5Centro Nacional de Biotecnología, C.S.I.C.; 6Department of Biomedical Sciences, Universidad de Alcalá, Spain; 7Centro de Investigaciones Biológicas, CSIC dperezsala@cib.csic.es.
Aldose reductase (AKR1B1) is a critical drug target due to its involvement in diabetic complications, inflammation and tumorigenesis. However, to date, development of clinically useful inhibitors has been largely unsuccessful. Cyclopentenone prostaglandins (cyPGs) are reactive lipid mediators that bind covalently to proteins and exert anti-inflammatory and antiproliferative effects in numerous settings. By pursuing the targets for modification by cyPGs we have found that the cyPG PGA1 binds to and inactivates AKR1B1. A PGA1-AKR1B1 adduct was observed, both by MALDI-TOF MS and by SDS-PAGE using biotinylated PGA1 (PGA1-B). Insight into the molecular interactions between AKR1B1 and PGA1 was advanced by molecular modeling. This anticipated the addition of PGA1 to active site Cys298 and the potential reversibility of the adduct, which was supported experimentally. Indeed, loss of biotin label from the AKR1B1-PGA1-B adduct was favored by glutathione, indicating a retro-Michael reaction, which unveils new implications of cyPG-protein interaction. PGA1 elicited only marginal inhibition of aldehyde reductase (AKR1A1), considered responsible for the severe adverse effects of many AKR1B1 inhibitors. Interestingly, other PG, including non-electrophilic PGE1 and PGE2, currently used in the clinical practice, inhibited the enzyme. Moreover, both PGA1 and PGE1 reduced the formation of sorbitol in an ex-vivo model of diabetic cataract to extents comparable to those
attained by the known AKR inhibitor epalrestat. Taken together, these results highlight the role of PG as AKR1B1 inhibitors and the interest of PG-related molecules as leads for the development of novel pharmacological tools.
PMID: 26487510 10.1124/mol.115.100693
Restoration of Microtubule Interaction and Cytotoxicity in D-seco Taxanes upon Incorporation of 20-Hydroxymethyl-4-allyloxy Groups.
Wang SR1, Yang CG1, Sánchez-Murcia PA2, Snyder JP3, Yan N1, Sáez-Calvo G4, Díaz JF4, Gago F2, Fang WS1.
Author information: 1State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College , 2A Nanwei Road, Beijing 100050, China. 2Área de Farmacología, Departamento de Ciencias Biomédicas, Universidad de Alcalá, E-28871 Alcalá de Henares, Unidad Asociada al Instituto de Química Médica del CSIC, E-28871 Madrid, Spain. 3Department of Chemistry, Emory University , 1515 Dickey Drive, Atlanta, Georgia 30322, United States. 4Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas , Ramiro de Maeztu 9, 28040 Madrid, Spain.
To probe the exact role of the oxetane D ring in both tubulin binding and cytotoxicity of taxanes, novel D-seco taxanes bearing a C4 ether substituent have been prepared from paclitaxel 1a. Among them, 20-hydroxymethyl-4-allyloxy D-seco taxane 5e is the most active in both tubulin and cytotoxicity assays. It is only slightly less potent than 1a on tubulin polymerization promotion in vitro and the most cytotoxic among all D-seco taxanes known to date. The reason for the loss and restoration of bioactivity for these D-seco taxanes is also discussed with the assistance of NMR and molecular modeling studies. From these results, we draw a conclusion that the intact D ring of taxanes is not strictly necessary for their binding to tubulin and cytotoxic effects.
PMID: 26649936 10.1039/C5OB02131F
A novel C,D-spirodioxene taxoid synthesized through an unexpected Pd-mediated ring cyclization.
Wang SR1, Sánchez-Murcia PA2, Gago F2, Fang WS1.
Author information: 1State Key Laboratory of Bioactive Substances and Functions of Natural Medicines, Institute of Materia Medica, Chinese Academy of Medical Sciences & Peking Union Medical College, 2A Nanwei Road, Beijing 100050, China. wfang@imm.ac.cn. 2Área de Farmacología, Departamento de Ciencias Biomédicas, Universidad de Alcalá, E-28871 Alcalá de Henares, Unidad Asociada al Instituto de Química Médica del CSIC, E-28871 Madrid, Spain.
A novel C,D-spirodioxene taxoid (6) was prepared from paclitaxel (1a), with the key steps including an unexpected Pd-mediated ring cyclization. The anti-tubulin activity of 6 was decreased relative to that of 1a and a previously reported C,D-spirolactone taxane (5). These observations could be rationalized on the basis of molecular modeling results. To the best of our knowledge, this is the first example indicating that 1,4-dioxenes can be synthesized from a mono-allyl vicinal diol through a Wacker-type cyclization. This strategy may be applicable to the synthesis of other C,D-spiro taxoids.
PMID: 26603551 10.1039/C5OB02131F
Conservation of antiviral activity and improved selectivity in PMEO-DAPym upon pyrimidine to triazine scaffold hopping.
Castro S1, Fernández-Cureses G1, Andrei G2, Snoeck R2, Sánchez-Murcia PA3, Korba B4, Gago F3, Balzarini J2, Camarasa MJ5.
Author information: 1Instituto de Química Médica (IQM-CSIC), Juan de la Cierva 3, E-28006 Madrid, Spain. 2Rega Institute for Medical Research, KU Leuven, Minderbroedersstraat 10, B-3000 Leuven, Belgium. 3Department of Biomedical Sciences, University of Alcalá, Unidad Asociada CSIC, E-28871 Alcalá de Henares, Madrid, Spain. 4Department of Microbiology and Immunology. Georgetown University Medical Center, 3900 Reservoir Road, Medical-Dental Building, Washington DC 200057, United States. 5Instituto de Química Médica (IQM-CSIC), Juan de la Cierva 3, E-28006 Madrid, Spain. Electronic address: mj.camarasa@iqm.csic.es.
Acyclic nucleoside phosphonates incorporating 2,4-diaminotriazine (DAT) as a 5-aza-analogue of the 2,4-diamino-pyrimidine (DAPym) nucleobase present in PMEO-DAPyms have been synthesized. The lead PMEO-DAT is as inhibitory against HIV, HBV, MSV and VZV replication as the parent PMEO-DAPym and equally inefficient at markedly affecting replication of HSV-1, HSV-2 and HCMV. A rationale for this similar biological profile is proposed on the basis of structural differences in the active site of the viral DNA polymerases. PMEO-DAT is, however, more selective because, unlike PMEO-DAPym, it does not stimulate secretion of β-chemokines in cultured PBMC.
PMID: 26275802 10.1016/j.antiviral.2015.08.006
Enantioselective oxidation of galactitol 1-phosphate by galactitol-1-phosphate 5-dehydrogenase from Escherichia coli.
Benavente R1, Esteban-Torres M2, Kohring GW3, Cortés-Cabrera Á4, Sánchez-Murcia PA4, Gago F4, Acebrón I1, de Las Rivas B2, Muñoz R2, Mancheño JM1.
1Department of Crystallography and Structural Biology, Institute of Physical Chemistry Rocasolano, CSIC, Serrano 119, 28006 Madrid, Spain. 2Laboratory of Bacterial Biotechnology, Institute of Food Science and Technology and Nutrition (ICTAN), CSIC, Juan de la Cierva 3, 28006 Madrid, Spain. 3Microbiology, Saarland University, Campus Gebäude A1.5, 66123 Saarbruecken, Germany. 4Department of Biomedical Sciences, School of Medicine and Health Sciences, University of Alcalá, 28871 Alcalá de Henares, Spain.
Galactitol-1-phosphate 5-dehydrogenase (GPDH) is a polyol dehydrogenase that belongs to the medium-chain dehydrogenase/reductase (MDR) superfamily. It catalyses the Zn2+- and NAD+-dependent stereoselective dehydrogenation of L-galactitol 1-phosphate to D-tagatose 6-phosphate. Here, three crystal structures of GPDH from Escherichia coli are reported: that of the open state of GPDH with Zn2+ in the catalytic site and those of the closed state in complex with the polyols Tris and glycerol, respectively. The closed state of GPDH reveals no bound cofactor, which is at variance with the conformational transition of the prototypical mammalian liver alcohol dehydrogenase. The main intersubunit-contacting interface within the GPDH homodimer presents a large internal cavity that probably facilitates the relative movement between the subunits. The substrate analogue glycerol bound within the active site partially mimics the catalytically relevant backbone of galactitol 1-phosphate. The glycerol binding mode reveals, for the first time in the polyol dehydrogenases, a pentacoordinated zinc ion in complex with a polyol and also a strong hydrogen bond between the primary hydroxyl group and the conserved Glu144, an interaction originally proposed more than thirty years ago that supports a catalytic role for this acidic residue.
PMID: 26143925 10.1107/S1399004715009281
A Computational Fragment-Based De Novo Design Protocol Guided by Ligand Efficiency Indices (LEI).
Cortés-Cabrera Á1, Gago F, Morreale A.
1Unidad de Bioinformática, Centro de Biología Molecular Severo Ochoa (CSIC-UAM), Campus de Cantoblanco UAM, Madrid, Spain.
We present a new protocol aimed at the structure-based design of drug-like molecules using a fragment approach. It starts from a suitably placed and well-defined "base fragment" and then uses an incremental construction algorithm and a scoring function to grow the molecule into prioritized candidates. The selection of the most promising solutions for synthesis and validation is guided by the optimization of the calculated ligand efficiency indices known as binding efficiency index (BEI) and surface efficiency index (SEI), which allow the user to navigate proficiently in chemico-biological space. A test case for the protocol is exemplified here using published data for inhibitors of protein kinase B, aka AKT, a key enzyme in several signal transduction pathways. Our procedure was able to identify the main features responsible for the binding of inhibitors and guided the selection process towards molecules that included or resembled those shown as the most active in the original studies.
PMID: 25709035 10.1007/978-1-4939-2486-8_8
Synthesis and evaluation of hybrid molecules targeting the vinca domain of tubulin.
Gherbovet O1, Sánchez-Murcia PA, García-Alvarez MC, Bignon J, Thoret S, Gago F, Roussi F.
Author information: 1Centre de Recherche de Gif, Institut de Chimie des Substances Naturelles, UPR 2301 du CNRS, Avenue de la Terrasse, 91198, Gif-sur-Yvette Cedex, France. fanny.roussi@cnrs.fr.
Some hybrids of vinca alkaloids and phomopsin A, linked by a glycine pattern, have been synthesized in one or two steps, by an insertion reaction and shown to inhibit microtubule assembly. These compounds have been elaborated in order to interact with both the "vinca site" and the "peptide site" of the vinca domain in tubulin. Two out of three hybrids are potent inhibitors of microtubules assembly and they present good cytotoxicity against different cell lines. Molecular modelling studies show that they could bind, within the vinca domain, in similar spatial regions as those of vinca and phomopsin thanks to the flexibility provided by the glycine linker used to elaborate these hybrids.
PMID: 25634805 10.1039/C4OB02114B
The Pyrimidine Nucleoside Phosphorylase of Mycoplasma hyorhinis and How It May Affect Nucleoside-Based Therapy.
Vande Voorde J1, Liekens S, Gago F, Balzarini J.
Author information:
1 Rega Institute for Medical Research , KU Leuven , Leuven , Belgium.
Mycoplasmas are opportunistic parasites and some species are suggested to preferentially colonize tumor tissue in cancer patients. We could demonstrate that the annotated thymidine phosphorylase (TP) gene in the genome of Mycoplasma hyorhinis encodes a pyrimidine nucleoside phosphorylase (PyNPHyor) that not only efficiently catalyzes thymidine but also uridine phosphorolysis. The kinetic characteristics of PyNPHyor-catalyzed nucleoside and nucleoside analogue (NA) phosphorolysis were determined. We demonstrated that the expression of such an enzyme in mycoplasma-infected cell cultures dramatically alters the activity of various anticancer/antiviral NAs such as 5-halogenated pyrimidine nucleosides, including 5-trifluorothymidine (TFT). Due to their close association with human
cancers, the presence of mycoplasmas may markedly influence the therapeutic efficiency of nucleoside-based drugs.
PMID: 24940697 10.1080/15257770.2013.851394
One-pot synthesis of Vinca Alkaloids-Phomopsin Hybrids.
Gherbovet O, Coderch C, Garcia Alvarez MC, Bignon J, Thoret S, Guéritte F, Gago F, Roussi F.
Hybrids of vinca alkaloids and phomopsin A have been elaborated with the aim of interfering with both the "vinca site" and the "peptide site" of the so-called vinca domain in tubulin. They were synthesized by an efficient one-pot procedure that links directly the octahydrophomopsin lateral chain to the velbenamine moiety of 7'-homo-anhydrovinblastine. In their modeled complexes with tubulin, these hybrids were found to superimpose nicely on the tubulin-bound structures of vinblastine and phomopsin A. This good matching can account for the fact that two of them are very potent inhibitors of microtubule assembly and display good cytotoxicity against four cancer cell lines.
PMID: 24871162 10.1021/jm500530v
Microwave-assisted synthesis of potent PDE7 inhibitors containing a thienopyrimidin-4-amine scaffold.
Sánchez AI1, Meneses R, Mínguez JM, Núñez A, Castillo RR, Filace F, Burgos C, Vaquero JJ, Alvarez-Builla J, Cortés-Cabrera A, Gago F, Terricabras E, Segarra V.
Author information:
1Departamento de Química Orgánica, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain. julio.alvarez@uah.es.
A series of novel thienopyrimidin-4-amines have been synthesized and evaluated as phosphodiesterase (PDE) inhibitors. A rationale for the observed selectivity against PDE7 has been obtained from molecular modelling studies on the most active compounds.
PMID: 24838636 10.1039/C4OB00175C
Esterase LpEst1 from Lactobacillus plantarum: A Novel and Atypical Member of the αβ Hydrolase Superfamily of Enzymes.
Alvarez Y1, Esteban-Torres M2, Cortés-Cabrera A3, Gago F3, Acebrón I4, Benavente R4, Mardo K5, de Las Rivas B2, Muñoz R2, Mancheño JM4.
Author information:
1Department of Crystallography and Structural Biology, Institute of Physical Chemistry Rocasolano, CSIC, Madrid, Spain; Center of Advanced Studies of Cuba, CITMA, Havana, Cuba.
2Laboratory of Bacterial Biotechnology, Institute of Food Science and Technology and Nutrition (ICTAN), CSIC, Madrid, Spain.
3Department of Biomedical Sciences, School of Medicine and Health Sciences, University of Alcalá, Alcalá de Henares, Madrid, Spain.
4Department of Crystallography and Structural Biology, Institute of Physical Chemistry Rocasolano, CSIC, Madrid, Spain.
5Institute of Molecular and Cell Biology, University of Tartu, Tartu, Estonia.
The genome of the lactic acid bacterium Lactobacillus plantarum WCFS1 reveals the presence of a rich repertoire of esterases and lipases highlighting their important role in cellular metabolism. Among them is the carboxylesterase LpEst1 a bacterial enzyme related to the mammalian hormone-sensitive lipase, which is known to play a central role in energy homeostasis. In this study, the crystal structure of LpEst1 has been determined at 2.05 Å resolution; it exhibits an αβ-hydrolase fold, consisting of a central β-sheet surrounded by α-helices, endowed with novel topological features. The structure reveals a dimeric assembly not comparable with any other enzyme from the bacterial hormone-sensitive lipase family, probably echoing the specific structural features of the participating subunits. Biophysical studies including analytical gel filtration and ultracentrifugation support the dimeric nature of LpEst1. Structural and mutational analyses of the substrate-binding pocket and active site together with biochemical studies provided insights for understanding the substrate profile of LpEst1 and suggested for the first time the conserved Asp173, which is adjacent to the nucleophile, as a key element in the stabilization of the loop where the oxyanion hole resides.
PMID: 24663330 10.1371/journal.pone.0092257
Structures reported by this article: 4C87 | , 4C88,4C89.
Leishmania infantum EndoG Is an Endo/Exo-Nuclease Essential for Parasite Survival.
Rico E1, Oliva C1, Gutierrez KJ1, Alzate JF2, Genes CM1, Moreno D1, Casanova E3, Gigante A3, Pérez-Pérez MJ3, Camarasa MJ3, Clos J4, Gago F5, Jiménez-Ruiz A1.
Author information:
1Departamento de Biología de Sistemas-Unidad Asociada al Consejo Superior de Investigaciones Científicas (CSIC), Universidad de Alcalá, Alcalá de Henares,
Madrid, Spain.
2Departamento de Microbiología y Parasitología, Facultad de Medicina, Universidad de Antioquia, Medellín, Colombia.
3Instituto de Química Médica - Consejo Superior de Investigaciones Científicas (IQM-CSIC), Madrid, Spain.
4Bernhard Nocht Institute for Tropical Medicine, Hamburg, Germany.
5Departamento de Ciencias Biomédicas - Unidad Asociada al Consejo Superior de Investigaciones Científicas (CSIC), Alcalá de Henares, Madrid, Spain.
EndoG, a member of the DNA/RNA non-specific ββα-metal family of nucleases, has been demonstrated to be present in many organisms, including Trypanosomatids. This nuclease participates in the apoptotic program in these parasites by migrating from the mitochondrion to the nucleus, where it takes part in the degradation of genomic DNA that characterizes this process. We now demonstrate that Leishmania infantum EndoG (LiEndoG) is an endo-exonuclease that has a preferential 5' exonuclease activity on linear DNA. Regardless of its role during apoptotic cell death, this enzyme seems to be necessary during normal development of the parasites as indicated by the reduced growth rates observed in LiEndoG hemi-knockouts and their poor infectivity in differentiated THP-1 cells. The pro-life role of this protein is also corroborated by the higher survival rates of parasites that over-express this protein after treatment with the LiEndoG inhibitor Lei49. Taken together, our results demonstrate that this enzyme plays essential roles in both survival and death of Leishmania parasites.
PMID: 24651293 10.1371/journal.pone.0089526
The molecular recognition of epothilones by microtubules and tubulin dimers revealed by biochemical and NMR approaches.
Canales A, Nieto L, Rodríguez-Salarichs J, Sánchez-Murcia PA, Coderch C, Cabrera AC, Paterson I, Carlomagno T, Gago F, Andreu JM, Altmann KH, Jiménez-Barbero J, Díaz JF.
The binding of epothilones to dimeric tubulin and to microtubules has been studied by means of biochemical and NMR techniques. We have determined the binding constants of epothilone A (EpoA) and B (EpoB) to dimeric tubulin, which are four orders of magnitude lower than those for microtubules, and we have elucidated the conformation and binding epitopes of EpoA and EpoB when bound to tubulin dimers and microtubules in solution. The determined conformation of epothilones when bound to dimeric tubulin is similar to that found by X-ray crystallographic techniques for the binding of EpoA to the Tubulin/RB3/TTL complex; it is markedly different from that reported for EpoA bound to zinc-induced sheets obtained by electron crystallography. Likewise, only the X-ray structure of EpoA bound to the Tubulin/RB3/TTL complex at the luminal site, but not the electron crystallography structure, is compatible with the results obtained by STD on the binding epitope of EpoA bound to dimeric tubulin, thus confirming that the allosteric change (structuring of the M-loop) is the biochemical mechanism of induction of tubulin assembly by epothilones. TR-NOESY signals of EpoA bound to microtubules have been obtained supporting the interaction with a transient binding site with a fast exchange rate (pore site), consistent with the notion that epothilones access the luminal site through the pore site, as has also been observed for taxanes. Finally, the differences in the tubulin binding affinities of a series of epothilone analogs has been
quantitatively explained using the newly determined binding pose and the COMBINE methodology.
PMID: 24524625 10.1021/cb400673h
Mechanistic insight into the reaction catalysed by bacterial type II dehydroquinases.
Coderch C, Lence E, Peón A, Lamb H, Hawkins AR, Gago F, González-Bello C.
The type II dehydroquinase (DHQ2), which is an essential enzyme in Helicobacter pylori and Mycobacterium tuberculosis, is recognized to be an attractive target for the development of new antibacterial agents. Computational and biochemical studies that help understand in atomic detail the catalytic mechanism of these bacterial enzymes are reported. Asp89*/Asp88* from a symmetry-related neighboring enzyme subunit proved to be the residue responsible for the deprotonation of the essential tyrosine to afford the catalytic tyrosinate, which triggers the enzymatic process. The essentiality of this residue is supported by results from site-directed mutagenesis. For H. pylori DHQ2, this reaction takes place through the assistance of a water molecule, while for M. tuberculosis DHQ2, the tyrosine is directly deprotonated by the aspartate residue. The participation of a water molecule in this deprotonation reaction is supported by solvent isotope effects and proton inventory studies. Results from molecular dynamics simulations provide details of the required motions for the catalytic turnover and a complete overview of the catalytic cycle. The product is expelled from the active site by the essential arginine and after a large conformational change of a loop containing two conserved arginines (Arg109/Arg108 and Arg113/Arg112), which reveals a previously unknown key role of these residues. The new insights can be used to advantage in the structure-based design of novel inhibitors.
PMID: 24392963 10.1042/BJ20131103
[Online data]
ALFA: Automatic Ligand Flexibility Assignment.
Klett J, Cortés-Cabrera A, Gil-Redondo R, Gago F, Morreale A.
ALFA is a fast computational tool for the conformational analysis of small molecules that uses a custom-made iterative algorithm to provide a set of representative conformers in an attempt to reproduce the diversity of states in which small molecules can exist, either isolated in solution or bound to a target. The results shown in this work prove that ALFA is fast enough to be integrated into massive high-throughput virtual screening protocols with the aim of incorporating ligand flexibility and also that ALFA reproduces crystallographic X-ray structures of bound ligands with great accuracy. Furthermore, the application includes a graphical user interface that allows its use through the popular molecular graphics program PyMOL to make it accessible to non-expert users. ALFA is distributed free of charge upon request from the authors.
PMID: 24392957 10.1021/ci400453n
New horizons in pharmaceutical biotechnology by melding biology and engineering.
Parayil A, Gago F.
Manus Biosynthesis, Cambridge, MA 02139, USA.
PMID: 24161714 10.1016/j.copbio.2013.10.001
Inhibitory effects of marine-derived DNA-binding antitumor tetrahydroisoquinolines on the Fanconi anemia pathway.
Martínez S, Pérez L, Galmarini CM, Aracil M, Tercero JC, Gago F, Albella B, Bueren JA.
Hematopoietic Innovative Therapies Division, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas (CIEMAT) and Centro de Investigación Biomédica en Red de Enfermedades Raras (CIBERER), E-28040 Madrid, Spain; Pharmamar S.A., Avda. de los Reyes, 1 - Pol. Ind. La Mina, E-28770 Colmenar Viejo, Madrid, Spain.
BACKGROUND AND PURPOSE: We have previously shown that cells deficient in the Fanconi anemia (FA) pathway are hypersensitive to trabectedin, a DNA-binding anticancer tetrahydroisoquinoline (DBAT) whose adducts functionally mimic a DNA inter-strand crosslink (ICL). Now we expand our observations to new DBATs and investigate whether our findings in primary untransformed cells can be reproduced in human cancer cells.
EXPERIMENTAL APPROACH: The sensitivity of FA-competent and FA-deficient transformed and untransformed cells to mitomycin C (MMC) and to three DBATs, trabectedin, Zalypsis and PM01183, was first assessed. Additionally, the functional interaction of these drugs with the FA pathway was comparatively investigated.
KEY RESULTS: While untransformed FA-deficient hematopoietic cells were hypersensitive to both MMC and DBATs, the response of FA-deficient squamous cell carcinoma (SCC) cells to DBATs was similar to that of their respective FA-competent counterparts, even though these FA-deficient SCC cells showed the expected hypersensitivity to MMC. Furthermore, while MMC always activated the FA pathway, DBATs inhibited FA pathway in the cancer cell lines tested and this enhanced their response to MMC.
CONCLUSIONS AND IMPLICATIONS: Taken together, our data show that although DBATs may functionally interact with DNA like agents that generate classical ICL, these drugs should be considered as FA pathway inhibitors, rather than activators. Moreover, this effect was most significant in a variety of cancer cells. We propose that the inhibitory effects of DBATs on the FA pathway could be exploited clinically with the aim of "fanconizing" cancer cells in order to make them more sensitive to other antitumor drugs.
PMID: 23937566 10.1111/bph.12331
Comparison of ultra-fast 2D and 3D ligand and target descriptors for side effect prediction and network analysis in polypharmacology.
Cortés-Cabrera A, Morris GM, Finn PW, Morreale A, Gago F.
Unidad de Bioinformática, Centro de Biología Molecular Severo Ochoa (CSIC/UAM), Campus de Cantoblanco, E-28049, Madrid, Spain; Departamento de Ciencias Biomédicas, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
BACKGROUND AND PURPOSE: Some computational methods currently exist that are employed to infer protein targets of small molecules and can therefore be used to find new targets for existing drugs, with the goals of repositioning the molecule for a different therapeutic purpose or explaining off-target effects due to multiple targeting. Inherent limitations, however, arise from the fact that chemical analogy is calculated on the basis of common frameworks or scaffolds and also because target information is neglected. The method we present addresses
these issues by taking into account 3D information from both the ligand and the target.
EXPERIMENTAL APPROACH: ElectroShape is an established method for ultra-fast comparison of the shapes and charge distributions of ligands that is validated here for prediction of on-target activities, off-target profiles and adverse effects of drugs and drug-like molecules mined from the DrugBank database.
KEY RESULTS: The method is shown to predict polypharmacology profiles and relate targets from two complementary viewpoints (ligand- and target-based networks).
CONCLUSIONS AND IMPLICATIONS: The open-access web tool presented here (http://ub.cbm.uam.es/chemogenomics/) allows interactive navigation in a unified 'pharmacological space' from the viewpoints of both ligands and targets. It also enables prediction of pharmacological profiles, including likely side effects, for new compounds. We hope this web interface will help many pharmacologists to become aware of this new paradigm (up to now mostly used in the realm of the so-called "chemical biology") and encourage its use with a view to revealing 'hidden' relationships between new and existing compounds and pharmacologically relevant targets.
PMID: 23826885 10.1111/bph.12294
Synthesis and Biological Evaluation of a New Series of Highly Functionalized 7'-Homo-Anhydrovinblastine Derivatives.
Gherbovet O, Coderch C, Garcia Alvarez MC, Bignon J, Thoret S, Martin MT, Guéritte F, Gago F, Roussi F.
Sixteen new 7' homo-anhydrovinblastine derivatives were prepared in one or two steps from vinorelbine by means of an original and regiospecific rearrangement and subsequent diastereoselective reduction. This strategy has allowed fast access to a family of vinca alkaloid derivatives with an enlarged and functionalized ring C'. Their synthesis and biological evaluation are reported. One compound (compound 35) is 1.7 times more active than vinorelbine as a tubulin assembly inhibitor. Moreover, some of these compounds are highly cytotoxic and two of them are more potent than vinorelbine on HCT116 and K562 cell lines. Molecular modeling studies, carried out with two of the new vinca derivatives, provide useful hints about how a given functionalization introduced at positions 7' and 8' of the C' ring results in improved binding interactions between one of the new derivatives and the interdimer interface when compared to the parent compound vinblastine.
PMID: 23822556 10.1021/jm4004347
Probing the Dimerization Interface of Leishmania infantum Trypanothione Reductase with Site-Directed Mutagenesis and Short Peptides.
Toro MA, Sánchez-Murcia PA, Moreno D, Ruiz-Santaquiteria M, Alzate JF, Negri A, Camarasa MJ, Gago F, Velázquez S, Jiménez-Ruiz A.
Departamento de Biología de Sistemas, Unidad Asociada de I+D+I al CSIC, Universidad de Alcalá, 28871 Alcalá de Henares, Madrid (Spain).
Binding at the interface: We tested the inhibitory activity of a set of peptide sequences derived from an α-helix of the dimeric trypanothione reductase from Leishmania infantum. Replacement of a glutamic acid residue with a lysine promoted monomer dissociation and enzyme inhibition.
© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
PMID: 23744811 10.1002/cbic.201200744
[Supporting Information]
Related article:
Sánchez-Murcia, A.; Ruiz-Santaquiteria, M.; Toro, M.A.; de Lucio, H.; Jiménez, M.A.; Gago, F.; Jiménez-Ruiz, A.; Camarasa, M.J.; Velázquez, S. "Comparison of hydrocarbon-and lactam-bridged cyclic peptides as dimerization inhibitors of Leishmania infantum trypanothione reductase"
RSC Advances, 5, 55784-55794 (2015) 10.1039/C5RA06853C
Antiviral agents: structural basis of action and rational design.
Menéndez-Arias L, Gago F.
Centro de Biología Molecular "Severo Ochoa" (Consejo Superior de Investigaciones Científicas & Universidad Autónoma de Madrid), c/ Nicolás Cabrera 1, Campus de Cantoblanco, E-28049 Madrid, Spain, lmenendez@cbm.uam.es.
During the last 30 years, significant progress has been made in the development of novel antiviral drugs, mainly crystallizing in the establishment of potent antiretroviral therapies and the approval of drugs inhibiting hepatitis C virus replication. Although major targets of antiviral intervention involve intracellular processes required for the synthesis of viral proteins and nucleic acids, a number of inhibitors blocking virus assembly, budding, maturation, entry or uncoating act on virions or viral capsids. In this review, we focus on the drug discovery process while presenting the currently used methodologies to identify novel antiviral drugs by using a computer-based approach. We provide examples illustrating structure-based antiviral drug development, specifically neuraminidase inhibitors against influenza virus (e.g. oseltamivir and zanamivir) and human immunodeficiency virus type 1 protease inhibitors (i.e. the development of darunavir from early peptidomimetic compounds such as saquinavir). A number of drugs in preclinical development acting against picornaviruses, hepatitis B virus and human immunodeficiency virus and their mechanism of action are presented to show how viral capsids can be exploited as targets of antiviral therapy.
PMID: 23737066 10.1007/978-94-007-6552-8_20.
5'-Trityl-Substituted Thymidine Derivatives as a Novel Class of Antileishmanial Agents: Leishmania infantum EndoG as a Potential Target.
Casanova E, Moreno D, Gigante A, Rico E, Genes CM, Oliva C, Camarasa MJ, Gago F, Jiménez-Ruiz A, Pérez-Pérez MJ.
Instituto de Química Médica (IQM-CSIC), Juan de la Cierva 3, 28006, Madrid (Spain).
Two series of 5'-triphenylmethyl (trityl)-substituted thymidine derivatives were synthesized and tested against Leishmania infantum axenic promastigotes and amastigotes. Several of these compounds show significant antileishmanial activity, with IC50 values in the low micromolar range. Among these, 3'-O-(isoleucylisoleucyl)-5'-O-(3,3,3-triphenylpropanoyl)thymidine displays particularly good activity against intracellular amastigotes. Assays performed to characterize the nature of parasite cell death in the presence of the
tritylthymidines indicated significant alterations in mitochondrial transmembrane potential, an increase in superoxide concentrations, and also significant decreases in DNA degradation during the cell death process. Results point to the mitochondrial nuclease LiEndoG as a target for the action of this family of compounds.
© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
PMID: 23625887 10.1002/cmdc.201300129
A structure-based design of new C2- and C13-substituted taxanes: tubulin binding affinities and extended quantitative structure-activity relationships using comparative binding energy (COMBINE) analysis.
Coderch C, Tang Y, Klett J, Zhang SE, Ma YT, Shaorong W, Matesanz R, Pera B, Canales A, Jiménez-Barbero J, Morreale A, Díaz JF, Fang WS, Gago F.
Área de Farmacología, Departamento de Ciencias Biomédicas, Universidad de Alcalá, E-28871 Alcalá de Henares, Unidad Asociada al Instituto de Química Médica del CSIC, Madrid, Spain. federico.gago@uah.es.
Ten novel taxanes bearing modifications at the C2 and C13 positions of the baccatin core have been synthesized and their binding affinities for mammalian tubulin have been experimentally measured. The design strategy was guided by (i) calculation of interaction energy maps with carbon, nitrogen and oxygen probes within the taxane-binding site of β-tubulin, and (ii) the prospective use of a structure-based QSAR (COMBINE) model derived from an earlier series comprising 47 congeneric taxanes. The tubulin-binding affinity displayed by one of the new compounds (CTX63) proved to be higher than that of docetaxel, and an updated COMBINE model provided a good correlation between the experimental binding free energies and a set of weighted residue-based ligand-receptor interaction energies for 54 out of the 57 compounds studied. The remaining three outliers from the original training series have in common a large unfavourable entropic contribution to the binding free energy that we attribute to taxane preorganization in aqueous solution in a conformation different from that compatible with tubulin binding. Support for this proposal was obtained from solution NMR experiments and molecular dynamics simulations in explicit water. Our results shed additional light on the determinants of tubulin-binding affinity for this important class of antitumour agents and pave the way for further rational structural modifications.
PMID: 23532250 10.1039/c3ob40407b
[Supporting Information PDF (1539K)]
Comparative Binding Energy (COMBINE) Analysis for Understanding the Binding Determinants of Type II Dehydroquinase Inhibitors.
Peón A, Coderch C, Gago F, González-Bello C.
Centro Singular de Investigación en Química Biológica y Materiales, Moleculares (CIQUS), Universidad de Santiago de Compostela calle Jenaro de la Fuente s/n, 15782 Santiago de Compostela (Spain).
Herein we report comparative binding energy (COMBINE) analyses to derive quantitative structure-activity relationship (QSAR) models that help rationalize the determinants of binding affinity for inhibitors of type II dehydroquinase (DHQ2), the third enzyme of the shikimic acid pathway. Independent COMBINE models were derived for Helicobacter pylori and Mycobacterium tuberculosis DHQ2, which is an essential enzyme in both these pathogenic bacteria that has no counterpart in human cells. These studies quantify the importance of the hydrogen bonding interactions between the ligands and the water molecule involved in the DHQ2 reaction mechanism. They also highlight important differences in the ligand interactions with the interface pocket close to the active site that could provide guides for future inhibitor design.
© 2013 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
PMID: 23450741 10.1002/cmdc.201200065
[Supporting Information]
Molecular simulations of drug-receptor complexes in anticancer research.
Gago F.
Department of Pharmacology, University of Alcalá, E-28871 Alcalá de Henares, Madrid, Spain. federico.gago@uah.es.
Molecular modeling and computer simulation techniques have matured significantly in recent years and proved their value in the study of drug-DNA, drug-DNA-protein, drug-protein and protein-protein interactions. Evolution in this area has gone hand-in-hand with an increased availability of structural data on biological macromolecules, major advances in molecular mechanics force fields and considerable improvements in computer technologies, most significantly processing speeds, multiprocessor programming and data-storage capacity. The information derived from molecular simulations of drug-receptor complexes can be used to extract structural and energetic information that is usually beyond current experimental possibilities, provide independent accounts of experimentally observed behavior, help in the interpretation of biochemical or pharmacological results, and open new avenues for research by posing novel relevant questions that can guide the design of new experiments. As drug-screening tools, ligand- and fragment-docking platforms stand out as powerful techniques that can provide candidate molecules for hit and lead
development. This review provides an overall perspective of the main methods and focuses on some selected applications to both classical and novel anticancer targets.
PMID: 23088276 10.4155/fmc.12.149
AtlasCBS: a web server to map and explore chemico-biological space.
Cortés-Cabrera A, Gago F, Morreale A, Abad-Zapatero C.
Departamento de Farmacología, Universidad de Alcalá, 28871 Alcalá de Henares, Madrid, Spain.
New approaches are needed that can help decrease the unsustainable failure in small-molecule drug discovery. Ligand Efficiency Indices (LEI) are making a great impact on early-stage compound selection and prioritization. Given a target-ligand database with chemical structures and associated biological affinities/activities for a target, the AtlasCBS server generates two-dimensional, dynamical, representations of its contents in terms of LEI. These variables allow an effective decoupling of the chemical (angular) and biological (radial) components. BindingDB, PDBBind and ChEMBL databases are currently implemented. Proprietary datasets can also be uploaded and compared. The utility of this atlas-like representation in the future of drug design is highlighted with some examples. The web server can be accessed at https://www.ebi.ac.uk/chembl/atlascbs/.
PMID: 22798082
CRDOCK: an ultrafast multipurpose protein-ligand docking tool.
Cortés-Cabrera A, Klett J, G Dos Santos H, Perona A, Gil-Redondo R, Francis SM, Priego EM, Gago F, Morreale A.
An ultrafast docking and virtual screening program, CRDOCK, is presented that contains (1) a searching engine that can use a variety of sampling methods and an initial energy evaluation function, (2) several energy minimization algorithms for fine-tuning the binding poses, and (3) different scoring functions. This modularity ensures the easy configuration of custom-made protocols that can be optimized depending on the problem in hand. CRDOCK employs a pre-computed library of ligand conformations that are initially generated from one-dimensional SMILES strings. Testing CRDOCK on two widely used benchmarks, the ASTEX diverse set and the Directory of Useful Decoys, yielded a success rate of ~75% in pose prediction and an average AUC of 0.66. A typical ligand can be docked, on average, in just ~13 seconds. Extension to a representative group of pharmacologically relevant G
protein-coupled receptors that have been recently co-crystallized with some selective ligands allowed us to demonstrate the utility of this tool and also to highlight some current limitations. CRDOCK is now included within VSDMIP, our integrated platform for drug discovery.
PMID: 22764680
Related article:
Klett, J.; Núñez-Salgado, A.; Dos Santos, H.G.; Cortés-Cabrera, A.; Perona, A.; Gil-Redondo, R.; Abia, D.; Gago, F.; Morreale, A. "MM-ISMSA: an ultra-fast and accurate scoring function for protein-protein docking"
Journal of Chemical Theory and Computation, 8, 3395-3408 (2012)
Introduction of a fluorine atom at C3 of 3-deazauridine shifts its antimetabolic activity from inhibition of CTP synthetase to inhibition of orotidylate decarboxylase, an early event in the de novo pyrimidine nucleotide biosynthesis pathway.
Balzarini J, Gago F, Kulik W, van Kuilenburg AB, Karlsson A, Peterson MA, Robins MJ.
Rega Institute for Medical Research, Belgium.
The antimetabolite prodrug 3-deazauridine (3DUrd) inhibits cytidine triphosphate (CTP) synthetase upon intracellular conversion to its triphosphate, which selectively depletes the intracellular CTP pools. Introduction of a fluorine atom at C3 of 3DUrd shifts its antimetabolic action to inhibition of the orotidylate (OMP) decarboxylase (ODC) activity of the uridylate (UMP) synthase enzyme complex that catalyzes an early event in pyrimidine nucleotide biosynthesis. This results in concomitant depletion of the intracellular UTP and CTP pools. The new prodrug (designated 3F-3DUrd) exerts its inhibitory activity as its monophosphate and is not further converted intracellularly to its triphosphate derivative to a detectable extent. Combinations with hypoxanthine and adenine markedly potentiate the cytostatic activity of 3F-3DUrd. This is likely due to depletion of PRPP (consumed in the HPRT/APRT reaction) and subsequent slowing of the PRPP-dependent OPRT reaction, which depletes OMP, the substrate for ODC. Further efficient anabolism by nucleotide kinases is compromised apparently due to the decrease in pKa brought about by the fluorine atom which affects the ionization state of the new prodrug. The 3F-3DUrd monophosphate exhibits new inhibitory properties against a different enzyme of the pyrimidine nucleotide metabolism, namely the ODC activity of UMP synthase.
PMID: 22730407
Recent Advances in Thymidine Kinase 2 (TK2) Inhibitors and New Perspectives for Potential Applications.
Priego EM, Karlsson A, Gago F, Camarasa MJ, Balzarini J, Pérez-Pérez MJ.
Instituto de Química Médica (C.S.I.C.), C/Juan de la Cierva, 3, E-28006 Madrid, Spain. empriego@iqm.csic.es.
Thymidine kinase 2 (TK2), encoded on chromosome 16q22 of the human genome, is a deoxynucleoside kinase (dNK) that catalyzes the phosphorylation of the pyrimidine deoxynucleosides 2'-deoxythymidine (dThd), 2'-deoxyuridine (dUrd) and 2'-deoxycytidine (dCyd) to the corresponding deoxynucleoside 5'-monophosphate derivatives. In contrast to the S-phase-specific thymidine kinase 1 (TK1), TK2 is constitutively expressed in the mitochondria and plays an important role in providing dNTPs for the replication and maintenance of mitochondrial DNA (mtDNA). The severe mitochondrial DNA depletion syndrome (MDS) has been associated with mutations in TK2, resulting in mtDNA depletion, isolated skeletal myopathy, and death of the individual at an early stage of life. Some antiviral nucleoside analogs, such as 3'-azido-dThd (AZT) that is targeting the human immunodeficiency virus (HIV)-encoded reverse transcriptase, are substrates for TK2 and it has been proposed that the mitochondrial toxicity observed after prolonged treatment with such drugs could be due to their interaction with TK2. Therefore, the design of specific TK2 inhibitors may be useful to investigate the role of TK2 in the maintenance and homeostasis of mitochondrial dNTP pools and its contribution to the mitochondrial toxicity of several antiviral and anticancer drugs. Since 2000,
several potent and selective TK2 inhibitors have been described. Besides bringing together previously reported inhibitors, special attention will be paid in this review to the new families of TK2 inhibitors more recently described, together with modeling studies and biological assays. Moreover, the last section will be focused on several recent investigations that suggest that depletion of mtDNA can take place both in tumorigenesis and during cancer treatment with certain nucleoside analogues.
PMID: 22571666
Characterization of pyrimidine nucleoside phosphorylase of Mycoplasma hyorhinis: implications for the clinical efficacy of nucleoside analogues.
Vande Voorde J, Gago F, Vrancken K, Liekens S, Balzarini J.
Rega Institute for Medical Research, Leuven, Belgium. jan.balzarini@rega.kuleuven.be
We demonstrate that the cytostatic and antiviral activity of pyrimidine nucleoside analogues is markedly decreased by a Mycoplasma hyorhinis infection and show that the phosphorolytic activity of the mycoplasmas is responsible for this. Since mycoplasmas are (i) an important cause of secondary infections in immunocompromised (e.g. HIV-infected) patients, and (ii) known to preferentially colonize tumor tissue in cancer patients, catabolic mycoplasma enzymes may compromise efficient chemotherapy of virus infections and cancer. In the genome of M. hyorhinis, a thymidine phosphorylase (TP) gene has been annotated. This gene was cloned, expressed in Escherichia coli and kinetically characterized. Whereas the mycoplasma TP efficiently catalyzes the phosphorolysis of thymidine (Km = 473 µM) and deoxyuridine (Km = 578 µM), it prefers uridine (Km = 92 µM) as a substrate. Our kinetic data and sequence analysis revealed that the annotated M. hyorhinis TP belongs to the nucleoside phosphorylase (NP)-II class pyrimidine nucleoside phosphorylases (PyNP), and is distinct from the NP-II class TP and NP-I class uridine phosphorylases (UP). M. hyorhinis PyNP also markedly differs from TP and UP in its substrate specificity towards therapeutic nucleoside analogues and susceptibility to clinically relevant drugs. Several kinetic properties of mycoplasma PyNP were explained by in silico analyses.
PMID: 22475552
Comparative Binding Energy (COMBINE) Analysis Supports a Proposal for the Binding Mode of Epothilones to β-Tubulin.
Coderch C, Klett J, Morreale A, Díaz JF, Gago F.
Departamento de Farmacología, Universidad de Alcalá, Edificio Medicina, Campus Universitario, 28871 Alcalá de Henares, Madrid (Spain).
The conformational preferences of epothilone A (EPA) and a 12,13-cyclopropyl C12-epimerized analogue were explored in aqueous solution using molecular dynamics simulations. The simulated conformers that provided an optimal fit in the paclitaxel binding site of mammalian β-tubulin were then selected. The resulting modeled complexes were simulated before and after refinement of the M-loop to improve the fitting and assess ligand stability within the binding pocket. The tubulin-bound conformation of EPA was found to be unlike a previously reported solution obtained through mixed crystallographic/NMR/modeling studies. However, our conformation was in agreement with an NMR-based proposal although the exact binding pose within the site was different. Molecular models were built for the complexes of 14 epothilone derivatives with β-tubulin. A projection to latent structures regression method succeeded in providing a good prediction of the experimentally measured binding enthalpies for the whole set of ligands by assigning weights to a selection of interaction energy terms. These receptor-based, quantitative structure-activity relationships support the proposed binding mode, help confirm and interpret previously acquired experimental data, shed additional light on the effect of several β-tubulin mutations on ligand binding, and can potentially direct further experimental studies.
© 2012 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
PMID: 22431398
[Supporting Information]
A reverse combination of structure-based and ligand-based strategies for virtual screening.
Cortés-Cabrera A, Gago F, Morreale A.
Departamento de Farmacología, Universidad de Alcalá, 28871 Alcalá de Henares, Madrid, Spain.
A new approach is presented that combines structure- and ligand-based virtual screening in a reverse way. Opposite to the majority of the methods, a docking protocol is first employed to prioritize small ligands ("fragments") that are subsequently used as queries to search for similar larger ligands in a database. For a given chemical library, a three-step strategy is followed consisting of (1) contraction into a representative, non-redundant, set of fragments, (2) selection of the three best-scoring fragments docking into a given macromolecular target site, and (3) expansion of the fragments' structures back into ligands by using them as queries to search the library by means of fingerprint descriptions and similarity criteria. We tested the performance of this approach on a collection of fragments and ligands found in the ZINC database and the directory of useful decoys, and compared the results with those obtained using a standard docking protocol. The new method provided better overall results and was several times faster. We also studied the chemical diversity that both methods cover using an in-house compound library and concluded that the novel approach performs similarly but at a much smaller computational cost.
PMID: 22395903
[Supporting Information]
Rationale for the opposite stereochemistry of the major monoadducts and interstrand crosslinks formed by mitomycin C and its decarbamoylated analogue at CpG steps in DNA and the effect of cytosine modification on reactivity.
Bueren-Calabuig JA, Negri A, Morreale A, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain. federico.gago@uah.es.
Mitomycin C (MMC) is a potent antitumour agent that forms a covalent bond with the 2-amino group of selected guanines in the minor groove of double-stranded DNA following intracellular reduction of its quinone ring and opening of its aziridine moiety. At some 5'-CG-3' (CpG) steps the resulting monofunctional adduct can evolve towards a more deleterious bifunctional lesion, which is known as an interstrand crosslink (ICL). MMC reactivity is enhanced when the cytosine bases are methylated (5 MC) and decreased when they are replaced with 5-F-cytosine (5FC) whereas the stereochemical preference of alkylation changes upon decarbamoylation. We have studied three duplex oligonucleotides of general formula d(CGATAAXGCTAACG) in which X stands for C, 5MC or 5FC. Using a combination of molecular dynamics simulations in aqueous solution, quantum mechanics and continuum electrostatics, we have been able to (i) obtain a large series of snapshots that facilitate an understanding in atomic detail of the distinct stereochemistry of monoadduct and ICL formation by MMC and its decarbamoylated analogue, (ii) provide an explanation for the altered reactivity of MMC towards DNA molecules containing 5MC or 5FC, and (iii) show the distinct accommodation in the DNA minor groove of the different covalent modifications, particularly the most cytotoxic C1α and C1β ICLs.
PMID: 22222915 10.1039/C1OB06675G
[Supporting Information]
Mechanistic Insight into the Catalytic Activity of ββα-Metallonucleases from Computer Simulations: Vibrio vulnificus Periplasmic Nuclease as a Test Case.
Bueren-Calabuig JA, Coderch C, Rico E, Jiménez-Ruiz A, Gago F.
Department of Pharmacology, Universidad de Alcalá, 28871 Alcalá de Henares, Madrid (Spain).
Using information from wild-type and mutant Vibrio vulnificus nuclease (Vvn) and I-PpoI homing endonuclease co-crystallized with different oligodeoxynucleotides, we have built the complex of Vvn with a DNA octamer and carried out a series of simulations to dissect the catalytic mechanism of this metallonuclease in a stepwise fashion. The distinct roles played in the reaction by individual active site residues, the metal cation and water molecules have been clarified by using a combination of classical molecular dynamics simulations and quantum mechanical calculations. Our results strongly support the most parsimonious catalytic mechanism, namely one in which a single water molecule from bulk solvent is used to cleave the phosphodiester bond and protonate the 3'-hydroxylate leaving group. © 2011 WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
PMID: 22114054
[Supporting
Information]
Tubulin-based Structure-affinity Relationships for Antimitotic Vinca Alkaloids.
Coderch C, Morreale A, Gago F.
federico.gago@uah.es.
The Vinca alkaloids are a group of widely used anticancer drugs, originally extracted from the Madagascar periwinkle, that disrupt microtubule dynamics in mammalian cells by interfering with proper assembly of α,β-tubulin heterodimers. They favor curved tubulin assemblies that destabilize microtubules and induce formation of spiral aggregates. Their binding energy profiles have been characterized by means of sedimentation velocity assays and the binding site of vinblastine at the interface between two tubulin dimers (α1β1-α2β2) has been ascertained by X-ray crystallographic studies on a complex of tubulin with the stathmin-like domain of protein RB3, albeit at relatively low resolution. Here we use molecular modeling and simulation techniques to build, refine and perform a comparative analysis of the three-dimensional complexes of vinblastine, vincristine, vinorelbine and vinflunine with a β1α2-tubulin interface in explicit water to rationalize the binding affinity differences in structural and energetic terms. Our results shed some more light into the binding determinants and the structure-activity relationships of these clinically useful agents.
PMID: 22044006
XPF-Dependent DNA Breaks and RNA Polymerase II Arrest Induced by Antitumor DNA Interstrand Crosslinking-Mimetic Alkaloids.
Feuerhahn S, Giraudon C, Martínez-Díez M, Bueren-Calabuig JA, Galmarini CM, Gago F, Egly JM.
Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS/INSERM/UdS, BP 163, 67404 Illkirch Cedex, C. U. Strasbourg, France.
Trabectedin and Zalypsis are two potent anticancer tetrahydroisoquinoline alkaloids that can form a covalent bond with the amino group of a guanine in selected triplets of DNA duplexes and eventually give rise to double-strand breaks. Using well-defined in vitro and in vivo assays, we show that the resulting DNA adducts stimulate, in a concentration-dependent manner, cleavage by the XPF/ERCC1 nuclease on the strand opposite to that bonded by the drug. They also inhibit RNA synthesis by: (1) preventing binding of transcription factors like Sp1 to DNA, and (2) arresting elongating RNA polymerase II at the same nucleotide position regardless of the strand they are located on. Structural models provide a rationale for these findings and highlight the similarity between this type of DNA modification and an interstrand crosslink.
PMID: 21867914
VSDMIP 1.5: an automated structure- and ligand-based virtual screening platform with a PyMOL graphical user interface.
Cabrera AC, Gil-Redondo R, Perona A, Gago F, Morreale A.
Departamento de Farmacología, Universidad de Alcalá, 28871, Alcalá de Henares, Madrid, Spain.
A graphical user interface (GUI) for our previously published virtual screening (VS) and data management platform VSDMIP (Gil-Redondo et al. J Comput Aided Mol Design, 23:171-184, 2009) that has been developed as a plugin for the popular molecular visualization program PyMOL is presented. In addition, a ligand-based VS module (LBVS) has been implemented that complements the already existing structure-based VS (SBVS) module and can be used in those cases where the receptor's 3D structure is not known or for pre-filtering purposes. This updated version of VSDMIP is placed in the context of similar available software and its LBVS and SBVS capabilities are tested here on a reduced set of the Directory of Useful Decoys database. Comparison of results from both approaches confirms the trend found in previous studies that LBVS outperforms SBVS. We also show that by combining LBVS and SBVS, and using a cluster of ~100 modern processors, it is possible to perform complete VS studies of several million molecules in less than a month. As the main processes in VSDMIP are 100% scalable, more powerful processors and larger clusters would notably decrease this time span. The plugin is distributed under an academic license upon request from the authors.
PMID: 21826555
Temperature-induced melting of double-stranded DNA in the absence and presence of covalently bonded antitumour drugs: insight from molecular dynamics simulations.
Bueren-Calabuig JA, Giraudon C, Galmarini CM, Egly JM, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, Institut de Génétique et de Biologie Moléculaire et Cellulaire, CNRS/INSERM/UdS, BP 163, 67404 Illkirch Cedex, C. U. Strasbourg, France and Cell Biology Department, PharmaMar, Avda. de los Reyes, 1 - Pol. Ind. La Mina, 28770
Colmenar Viejo, Madrid, Spain.
The difference in melting temperature of a double-stranded (ds) DNA molecule in the absence and presence of bound ligands can provide experimental information about the stabilization brought about by ligand binding. By simulating the dynamic behaviour of a duplex of sequence 5'-d(TAATAACGGATTATT)·5'-d(AATAATCCGTTATTA) in 0.1 M NaCl aqueous solution at 400 K, we have characterized in atomic detail its complete thermal denaturation profile in <200 ns. A striking asymmetry was observed on both sides of the central CGG triplet and the strand separation process was shown to be strongly affected by bonding in the minor groove of the prototypical interstrand crosslinker mitomycin C or the monofunctional tetrahydroisoquinolines trabectedin (Yondelis®), Zalypsis® and PM01183. Progressive helix unzipping was clearly interspersed with some reannealing events, which were most noticeable in the oligonucleotides containing the monoadducts, which maintained an average of 6 bp in the central region at the end of the simulations. These significant differences attest to the demonstrated ability of these drugs to stabilize dsDNA, stall replication and transcription forks, and recruit DNA repair proteins. This stabilization, quantified here in terms of undisrupted base pairs, supports the view that these monoadducts can functionally mimic a DNA interstrand crosslink.
PMID: 21727089 10.1093/nar/gkr512
Identification of Aldo-keto reductase AKR1B10 as a Selective Target for Modification and Inhibition by prostaglandin A1: Implications for Anti-tumoral Activity.
Díez-Dacal B, Gayarre J, Gharbi S, Timms JF, Coderch C, Gago F, Pérez-Sala D.
Chemical and Physical Biology, Centro de Investigaciones Biológicas, CSIC.
Cyclopentenone prostaglandins (cyPG) are reactive eicosanoids that may display anti-inflammatory and anti-proliferative actions, possibly offering therapeutic potential. Here we report the identification of members of the aldo-keto reductase (AKR) family as selective targets of the cyPG prostaglandin A1 (PGA1). AKR enzymes metabolize drugs containing aldehydes and other carbonyl groups and involved in inflammation and tumorigenesis. Thus, these enzymes represent a class of targets to develop small molecule inhibitors with therapeutic activity. Molecular modelling studies pointed to the covalent binding of PGA1 to Cys299, close to the active site of AKR, with His111 and Tyr49, which are highly conserved in the AKR family, playing a role in PGA1 orientation. Among AKR enzymes, AKR1B10 is considered as a tumor marker and contributes to tumor development and chemoresistance. We validated the direct modification of AKR1B10 by biotinylated PGA1 (PGA1-B) in cells, and confirmed that mutation of Cys299 abolishes PGA1-B incorporation, whereas substitution of His111 or Tyr49 reduced the interaction. Modification of AKR1B10 by PGA1 correlated with loss of enzymatic activity and both effects were increased by depletion of cellular glutathione. Moreover, in lung cancer cells PGA1 reduced tumorigenic potential and increased accumulation of the AKR substrate doxorubicin, potentiating cell cycle arrest induced by this chemotherapeutic agent. Our findings define PGA1 as a new AKR inhibitor and they offer a framework to develop compounds that could counteract cancer chemoresistance.
PMID: 21507934 10.1158/0008-5472.CAN-10-3816
Unusual Approach to 3-Aryl-2-aminopyridines through a Radical Mechanism: Synthesis and Theoretical Rationale from Quantum Mechanical Calculations.
Camacho-Artacho M, Abet V, Frutos LM, Gago F, Alvarez-Builla J, Burgos C.
Departamentos de Farmacología, Química Física y Química Orgánica, Universidad de Alcalá.
Tris(trimethylsilyl)silane and azobis(cyclohexanenitrile) promoted the easy intramolecular arylation of aryl bromopyridine carbamates through a radical [1,6] ipso substitution process. These substrates showed a preference for this type of reaction over the alternative [1,7] addition. The results were rationalized by making use of quantum mechanical calculations and computer graphics.
PMID: 21265527 10.1021/jo102122h
Synthesis, modeling and evaluation of 3'-(1-aryl-1H-tetrazol-5-ylamino)-substituted 3'-deoxythymidine derivatives as potent and selective human mitochondrial thymidine kinase inhibitors.
Van Poecke S, Negri A, Janssens J, Solaroli N, Karlsson A, Gago F, Balzarini J, Van Calenbergh S.
Laboratory for Medicinal Chemistry, Faculty of Pharmaceutical Sciences (FFW), Ghent University, Harelbekestraat 72, B-90000, Gent, Belgium. serge.vancalenbergh@ugent.be.
Based on the presumed binding mode of an earlier identified inhibitor, we herein report new 3'-modified nucleosides as potent and selective inhibitors of mitochondrial thymidine kinase (TK2). A series of thirteen 3'-amino-, 3'-guanidino- and 3'-tetrazole-containing nucleosides were synthesized and evaluated for their TK2 inhibitory activity. Within the tetrazole series, compounds with nanomolar inhibitory activity were identified. A homology model of TK2 allowed to elucidate the observed activities. Introduction of a 2-bromovinyl group on C-5 of the pyrimidine base of the most promising 3'-derivative further improved the inhibitory activity, and caused a significant increase in the selectivity for TK2 versus TK1. Interestingly, for the current series of analogues, a strong correlation was observed between TK2 and Drosophila melanogaster dNK inhibition, further substantiating the phylogenetic relationship between these two nucleoside kinases.
PMID: 21127790
PM01183, a new DNA minor groove covalent binder with potent in vitro and in vivo anti-tumour activity.
Leal J, Martínez-Díez M, García-Hernández V, Moneo V, Domingo A, Bueren-Calabuig J, Negri A, Gago F, Guillén-Navarro M, Avilés P, Cuevas C, García-Fernández L, Galmarini C
Cell Biology Department, Pharmamar SA, Avda. de los Reyes, 1, 28770 Colmenar Viejo, Madrid, Spain.
BACKGROUND AND PURPOSE: PM01183 is a new synthetic tetrahydroisoquinoline alkaloid that is currently in phase I clinical development for the treatment of solid tumours. In this study we have characterized the interactions of PM01183 with selected DNA molecules of defined sequence and its in vitro and in vivo cytotoxicity.
EXPERIMENTAL APPROACH: DNA binding characteristics of PM01183 were studied using electrophoretic mobility shift assays, fluorescence-based melting kinetic experiments and computational modelling methods. Its mechanism of action was investigated using flow cytometry, Western blot analysis and fluorescent microscopy. In vitro anti-tumour activity was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and the in vivo activity utilized several human cancer models.
KEY RESULTS: Electrophoretic mobility shift assays demonstrated that PM01183 bound to DNA. Fluorescence-based thermal denaturation experiments showed that the most favourable DNA triplets providing a central guanine for covalent adduct formation are AGC, CGG, AGG and TGG. These binding preferences could be rationalized using molecular modelling. PM01183-DNA adducts in living cells give rise to double-strand breaks, triggering S-phase accumulation and apoptosis. The potent cytotoxic activity of PM01183 was ascertained in a 23-cell line panel with a mean GI50 value of 2.7 nM. In four murine xenograft models of human cancer, PM01183 inhibited tumour growth significantly with no weight loss of treated animals.
CONCLUSIONS AND IMPLICATIONS: PM01183 is shown to bind to selected DNA sequences and to promote apoptosis by inducing double-strand breaks at nanomolar concentrations. The potent anti-tumour activity of PM01183 in several murine models of human cancer supports its development as a novel anti-neoplastic agent.
PMID: 20977459 10.1111/j.1476-5381.2010.00945.x
Understanding the Key Factors that Control the Inhibition of Type II Dehydroquinase by (2R)-2-Benzyl-3-dehydroquinic Acids.
Peón A, Otero JM, Tizón L, Prazeres VF, Llamas-Saiz AL, Fox GC, van Raaij MJ, Lamb H, Hawkins AR, Gago F, Castedo L, González-Bello C.
Departamento de Química Orgánica y Centro Singular de Investigación en Química Biológica y Materiales Moleculares, Universidad de Santiago de Compostela, calle Jenaro de la Fuente s/n, 15782 Santiago de Compostela (Spain), Fax: (+34)981-595012.
The binding mode of several substrate analogues, (2R)-2-benzyl-3-dehydroquinic acids 4, which are potent reversible competitive inhibitors of type II dehydroquinase (DHQ2), the third enzyme of the shikimic acid pathway, has been investigated by structural and computational studies. The crystal structures of Mycobacterium tuberculosi and Helicobacter pylori DHQ2 in complex with one of the most potent inhibitor, p-methoxybenzyl derivative 4 a, have been solved at 2.40 Å and 2.75 Å, respectively. This has allowed the resolution of the M. tuberculosis DHQ2 loop containing residues 20-25 for the first time. These structures show the key interactions of the aromatic ring in the active site of both enzymes and additionally reveal an important change in the conformation and flexibility of the loop that closes over substrate binding. The loop conformation and the binding mode of compounds 4 b-d has been also studied by molecular dynamics simulations, which suggest that the benzyl group of inhibitors 4 prevent appropriate orientation of the catalytic tyrosine of the loop for proton abstraction and disrupts its basicity.
PMID: 20815012
| |
Structures reported by this article: 2XB8 / 2XB9 |
3'-[4-Aryl-(1,2,3-triazol-1-yl)]-3'-deoxythymidine Analogues as Potent and Selective Inhibitors of Human Mitochondrial Thymidine Kinase.
Van Poecke S, Negri A, Gago F, Van Daele I, Solaroli N, Karlsson A, Balzarini J, Van Calenbergh S.
Laboratory for Medicinal Chemistry (FFW), Ghent University, Harelbekestraat 72, 9000 Gent, Belgium.
In an effort to increase the potency and selectivity of earlier identified substrate-based inhibitors of mitochondrial thymidine kinase 2 (TK-2), we now describe the synthesis of new thymidine analogues containing a 4- or 5-substituted 1,2,3-triazol-1-yl substituent at the 3'-position of the 2'-deoxyribofuranosyl ring. These analogues were prepared by Cu- and Ru-catalyzed cycloadditions of 3'-azido-3'-deoxythymidine and the appropriate alkynes, which produced the 1,4- and 1,5-triazoles, respectively. Selected analogues showed nanomolar inhibitory activity for TK-2, while virtually not affecting the TK-1 counterpart. Enzyme kinetics indicated a competitive and uncompetitive inhibition profile against thymidine and the cosubstrate ATP, respectively. This behavior is rationalized by suggesting that the inhibitors occupy the substrate-binding site in a TK-2-ATP complex that maintains the enzyme's active site in a closed conformation through the stabilization of a small lid domain.
PMID: 20218622
Intramolecular Cation-pi Interactions As the Driving Force To Restrict the Conformation of Certain Nucleosides.
Casanova E, Priego EM, Jimeno ML, Aguado L, Negri A, Gago F, Camarasa MJ, Pérez-Pérez MJ.
Instituto de Química Médica (CSIC), Juan de la Cierva 3, E-28006 Madrid, Spain.
Despite the well-established importance of intermolecular cation-pi interactions in molecular recognition, intramolecular cation-pi interactions have been less studied. Here we describe how the simultaneous presence of an aromatic ring at the 5'-position of an inosine derivative and a positively charged imidazolium ring in the purine base drive the conformation of the nucleoside toward a very major conformer in solution that is stabilized by an intramolecular cation-pi interaction. Therefore, the cation-pi interaction between imidazolium ions and aromatic rings can also be proposed in the design of small molecules where this type of interaction is desirable. The imidazolium ion can be obtained by a simple acidification of the pH of the media. So a simple change in pH can shift the conformational equilibrium from a random to a restricted conformation stabilized by an intramolecular cation-pi interaction. Thus the here described nucleosides can be considered as a new class of pH-dependent conformationally switchable molecules.
PMID: 20192191
gCOMBINE: A graphical user interface to perform structure-based comparative binding energy (COMBINE) analysis on a set of ligand-receptor complexes.
Gil-Redondo R, Klett J, Gago F, Morreale A.
Unidad de Bioinformática, Centro de Biología Molecular Severo Ochoa, Campus UAM, c/ Nicolás Cabrera 1, Madrid 28049, Spain.
We present gCOMBINE, a Java-written graphical user interface (GUI) for performing comparative binding energy (COMBINE) analysis (Ortiz et al. J Med Chem 1995; 38:2681-2691) on a set of ligand-receptor complexes with the aim of deriving highly informative quantitative structure-activity relationships. The essence of the method is to decompose the ligand-receptor interaction energies into a series of terms, explore the origins of the variance within the set using Principal Component Analysis, and then assign weights to selected ligand-residue interactions using partial least squares analysis to correlate with the experimental activities or binding affinities. The GUI allows plenty of interactivity and provides multiple plots representing the energy descriptors entering the analysis, scores, loadings, experimental versus predicted regression lines, and the evolution of parameters such as r2 (correlation coefficient), q2 (cross-validated r2), and prediction errors as the number of extracted latent variables increases. Other representative features include the implementation of a sigmoidal dielectric function for electrostatic energy calculations, alternative cross-validation procedures (leave-N-out and random groups), drawing of confidence ellipses, and the possibility to carry out several additional tasks such as optional truncation of positive interaction energy values and generation of ready-to-use PDB files containing information related to the importance for activity of individual protein residues. This information can be displayed and color-coded using a standard molecular graphics program such as PyMOL. It is expected that this user-friendly tool will expand the applicability of the COMBINE analysis method and encourage more groups to use it in their drug design research programs. Proteins 2009. c © 2009 Wiley-Liss, Inc.
PMID: 19705486
Protein-protein interactions at an enzyme-substrate interface: Characterization of transient reaction intermediates throughout a full catalytic cycle of Escherichia coli thioredoxin reductase.
Negri A, Rodríguez-Larrea D, Marco E, Jiménez-Ruiz A, Sánchez-Ruiz JM, Gago F.
Departmento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
A large collection of structural snapshots along a full catalytic cycle of Escherichia coli thioredoxin reductase (TrxR) has been generated and characterized using a combination of theoretical methods. Molecular models were built starting from the available X-ray crystallographic structures of dimeric wild-type TrxR in the flavin-oxidizing conformation and a C135S TrxR mutant enzyme in a flavin-reducing conformation "trapped" by a cross-link between Cys138 of TrxR and Cys32 of C35S mutant thioredoxin (Trx). The transition between these two extreme states, which is shown to be reproduced in a normal mode analysis, as well as natural cofactor binding and dissociation, were simulated for the wild-type species using unrestrained and targeted molecular dynamics following docking of oxidized Trx to reduced TrxR. The whole set of simulations provides a comprehensive structural framework for understanding the mechanism of disulfide reduction in atomic detail and identifying the most likely intermediates that facilitate entry of NADPH and exit of NADP+. The crucial role assigned to Arg73 and Lys36 of Trx in substrate binding and complex stabilization was ascertained when R73G, R73D, and K36A site-directed mutants of Trx were shown to be impaired to different extents in their ability to be reduced by TrxR. On the basis of previous findings and the results reported herein, E. coli TrxR appears as a beautifully engineered molecular machine that is capable of synchronizing cofactor capture and ejection with substrate binding and redox activity through an interdomain twisting motion. Proteins 2009. c © 2009 Wiley-Liss, Inc.
PMID: 19585660
[Supplementary Material]
The dual role of thymidine phosphorylase in cancer development and chemotherapy.
Bronckaers A, Gago F, Balzarini J, Liekens S.
Rega Institute for Medical Research, K.U.Leuven, B-3000 Leuven, Belgium.
Thymidine phosphorylase (TP), also known as "platelet-derived endothelial cell growth factor" (PD-ECGF), is an enzyme, which is upregulated in a wide variety of solid tumors including breast and colorectal cancers. TP promotes tumor growth and metastasis by preventing apoptosis and inducing angiogenesis. Elevated levels of TP are associated with tumor aggressiveness and poor prognosis. Therefore, TP inhibitors are synthesized in an attempt to prevent tumor angiogenesis and metastasis. TP is also indispensable for the activation of the extensively used 5-fluorouracil prodrug capecitabine, which is clinically used for the treatment of colon and breast cancer. Clinical trials that combine capecitabine with TP-inducing therapies (such as taxanes or radiotherapy) suggest that increasing TP expression is an adequate strategy to enhance the antitumoral efficacy of capecitabine. Thus, TP plays a dual role in cancer development and therapy: on the one hand, TP inhibitors can abrogate the tumorigenic and metastatic properties of TP; on the other, TP activity is necessary for the activation of several chemotherapeutic drugs. This duality illustrates the complexity of the role of TP in tumor progression and in the clinical response to fluoropyrimidine-based chemotherapy. c © 2009 Wiley Periodicals, Inc. Med Res Rev.
PMID: 19434693
Molecular pharmacology and antitumor activity of Zalypsis® in several human cancer cell lines.
Leal JF, García-Hernández V, Moneo V, Domingo A, Bueren-Calabuig JA, Negri A, Gago F, Guillén-Navarro MJ, Avilés P, Cuevas C, García-Fernández LF, Galmarini CM.
Cell Biology Department, Pharmamar SA, Avda. de los Reyes, 1, 28770 Colmenar Viejo, Madrid, Spain.
Zalypsis® is a new synthetic alkaloid tetrahydroisoquinoline antibiotic that has a reactive carbinolamine group. This functionality can lead to the formation of a covalent bond with the amino group of selected guanines in the DNA double helix, both in the absence and in the presence of methylated cytosines. The resulting complex is additionally stabilized by the establishment of one or more hydrogen bonds with adjacent nucleotides in the opposite strand as well as by van der Waals interactions within the minor groove. Fluorescence-based thermal denaturation experiments demonstrated that the most favorable DNA triplets for covalent adduct formation are AGG, GGC, AGC, CGG and TGG, and these preferences could be rationalized on the basis of molecular modeling results. Zalypsis®-DNA adducts eventually give rise to double-strand breaks, triggering S-phase accumulation and apoptotic cell death. The potent cytotoxic activity of Zalypsis® was ascertained in a 24 cell line panel. The mean IC50 value was 7 nM and leukemia and stomach tumor cell lines were amongst the most sensitive. Zalypsis® administration in four murine xenograft models of human cancer demonstrates significant tumor growth inhibition that is highest in the Hs746t gastric cancer cell line with no weight loss of treated animals. Taken together, these results indicate that the potent antitumor activity of Zalypsis® supports its current development in the clinic as an anticancer agent.
PMID: 19427997
Identification of aspartic acid-203 in human thymidine phosphorylase as an important residue for both catalysis and non-competitive inhibition by the small molecule "crystallization chaperone" 5'-O-tritylinosine (KIN59).
Bronckaers A, Aguado L, Negri A, Camarasa MJ, Balzarini J, Pérez-Pérez MJ, Gago F, Liekens S.
Rega Institute for Medical Research, K.U.Leuven, B-3000 Leuven, Belgium.
Thymidine phosphorylase (TP) is a catabolic enzyme in thymidine metabolism that is frequently upregulated in many solid tumors. Elevated TP levels are associated with tumor angiogenesis, metastasis and poor prognosis. Therefore, the use of TP inhibitors might offer a promising strategy for cancer treatment. The tritylated inosine derivative 5'-O-tritylinosine (previously designated KIN59) is a non-competitive inhibitor of TP which was previously found to be instrumental for the crystallization of human TP. A combination of computational studies including normal mode analysis, automated ligand docking and molecular dynamics simulations were performed to define a plausible binding site for 5'-O-tritylinosine on human TP. A cavity in which 5'-O-tritylinosine could fit was identified in the vicinity of the Gly405-Val419 loop at a distance of about 11A from the substrate-binding site. In the X-ray crystal structure, this pocket is characterized by an intricate hydrogen-bonding network in which Asp203 was found to play an important role to afford the loop stabilization that is required for efficient enzyme catalysis. Site-directed mutagenesis of this amino acid residue afforded a mutant enzyme with a severely compromised catalytic efficiency (Vmax/Km of mutant enzyme approximately 50-fold lower than for wild-type TP) and pronounced resistance to the inhibitory effect of 5'-O-tritylinosine. In contrast, the D203A mutant enzyme kept full sensitivity to the competitive inhibitors 6-aminothymine and 6-amino-5-bromouracil, which is in line with the kinetic properties of these inhibitors. Our findings reveal the existence of a previously unrecognized site of TP that can be targeted by small molecules to inhibit the catalytic activity of TP.
PMID: 19389384
Human Mitochondrial Thymidine Kinase (TK-2) is Selectively Inhibited by 3'-Thiourea Derivatives of β-Thymidine. Identification of residues crucial for both inhibition and catalytic activity.
Balzarini J, Van Daele I, Negri A, Solaroli N, Karlsson A, Liekens S, Gago F, Van Calenbergh S.
Rega Institute for Medical Research.
Substituted 3'-thiourea derivatives of β-thymidine (dThd) and 5'-thiourea derivatives of α-dThd have been evaluated for their inhibitory activity against recombinant human cytosolic dThd kinase-1 (TK-1), human mitochondrial TK-2, herpes simplex virus type 1 (HSV-1) TK and varicella-zoster virus (VZV) TK. Several substituted 3'-thiourea derivatives of β-dThd proved highly inhibitory to and selective for TK-2(IC50: 0.15-3.1 µM). The 3'-C-branched p-methylphenyl (1) and 3-CF3-4-Cl-phenyl (7) thiourea derivatives of β-dThd showed competitive inhibition of TK-2 when dThd was used as the variable substrate (Ki: 0.40 µM and 0.05 µM, respectively) but uncompetitive inhibition in the presence of variable concentrations of ATP (Ki: 15 µM and 2.0 µM, respectively). These kinetic properties of 1 and 7 against TK-2 could be accounted for by molecular modeling showing that two hydrogen bonds can be formed between the thiourea nitrogens of 7 and the oxygens of the γ-phosphate of ATP. The importance of several active-site residues was assessed by site-directed mutagenesis experiments on TK-2 and the related HSV-1 TK. The low Ki/Km ratios for 1 and 7 (0.38 and 0.039 against dThd, and 0.75 and 0.12 against ATP, respectively) indicate that these compounds are amongst the most potent inhibitors of TK-2 described so far. In addition, a striking close correlation was found between the inhibitory activities of the test compounds against TK-2 and Mycobacterium tuberculosis thymidylate kinase that is strongly indicative of close structural and/or functional similarities between both enzymes in relation to their mode of interaction with these nucleoside analogue inhibitors.
PMID: 19233899
Leishmania infantum expresses a mitochondrial nuclease homologous to EndoG that migrates to the nucleus in response to an apoptotic stimulus.
Rico E, Alzate JF, Arias AA, Moreno D, Clos J, Gago F, Moreno I, Domínguez M, Jiménez-Ruiz A.
Departamento de Bioquímica y Biología Molecular, Campus Universitario, Universidad de Alcalá, 28871 Alcalá de Henares, Madrid, Spain.
It is increasingly accepted that single-celled organisms, such as Leishmania parasites, are able to undergo a cell death process that resembles apoptosis in metazoans and is induced by a variety of stimuli. However, the molecular mechanisms that participate and regulate this death process are still very poorly described, and very few of the participating molecules have been identified. Because DNA degradation is probably the most frequently characterized event during programmed cell death in Leishmania parasites, we have focused on identifying a candidate nuclease responsible for this effect during the cell death process. The results presented herein demonstrate that Leishmania infantum promastigotes express a nuclease similar to the endonuclease G of higher eukaryotes which, according to its predicted structure, belongs to the ββα metal superfamily of nucleases. Its cation dependence resembles that of the EndoGs present in other organisms and, similarly to them, it is inhibited by moderate concentrations of K+ or Na+. L. infantum EndoG contains a signal peptide that causes its translocation to the mitochondrion where it is maintained under normal growth conditions. However, under the pressure of a death stimulus such as edelfosine treatment, L. infantum EndoG is released from the single mitochondrion and translocates to the nucleus, where it is thought to participate in the process of DNA degradation that is associated with programmed cell death. Our results also demonstrate that overexpression of the nuclease in edelfosine-treated promastigotes causes a significant increase in the percentage of TUNEL-positive parasites.
PMID: 18940204
The protein kinase inhibitor balanol: structure-activity relationships and structure-based computational studies.
Pande V, Ramos MJ, Gago F.
Departamento de Farmacología, Universidad de Alcalá, Alcalá de Henares, 28871 Madrid, Spain. federico.gago@uah.es.
Balanol, a fungal metabolite, is a potent ATP-competitive inhibitor of Protein Kinase C (PKC) and Protein Kinase A (PKA), important targets in oncology. Since its discovery in 1993, a number of studies have been performed in order to design selective and bioavailable balanol analogs. Several crystal structures of PKA in complex with balanol and a few analogs bound within the catalytic site have also been solved providing insight about the key interactions for binding. The PKA-balanol complex has also served as an interesting model system for structure-based ligand design and validation of a number of computational methodologies aimed at both understanding the physical basis for molecular recognition and addressing the important issue of protein flexibility in ligand binding. We provide an overview of the structure-activity relationships of balanol analogs and summarize the progress made in structural and computational studies involving balanol.
PMID: 18690827
Optimization of taxane binding to microtubules: binding affinity dissection and incremental construction of a high-affinity analog of Paclitaxel.
Matesanz R, Barasoain I, Yang CG, Wang L, Li X, de Inés C, Coderch C, Gago F, Barbero JJ, Andreu JM, Fang WS, Díaz JF.
Centro de Investigaciones Biológicas, Consejo Superior de Investigaciones Científicas, Ramiro de Maeztu 9, 28040 Madrid, Spain.
The microtubule binding affinities of a series of synthetic taxanes have been measured with the aims of dissecting individual group contributions and obtaining a rationale for the design of novel compounds with the ability to overcome drug resistance. As previously observed for epothilones, the positive and negative contributions of the different substituents to the binding free energies are cumulative. By combining the most favorable substitutions we increased the binding affinity of paclitaxel 500-fold. Insight into the structural basis for this improvement was gained with molecular modeling and NMR data obtained for microtubule-bound docetaxel. Taxanes with affinities for microtubules well above their affinities for P-glycoprotein are shown not to be affected by multidrug resistance. This finding strongly indicates that optimization of the ligand-target interaction is a good strategy to overcome multidrug resistance mediated by efflux pumps.
PMID: 18559268
Relevance of the Fanconi anemia pathway in the response of human cells to trabectedin.
Casado JA, Río P, Marco E, García-Hernández V, Domingo A, Pérez L, Tercero JC, Vaquero JJ, Albella B, Gago F, Bueren JA.
Division of Hematopoiesis and Gene Therapy Program, Centro de Investigaciones Energéticas, Medioambientales y Tecnológicas, Avenida Complutense 22, 28040 Madrid, Spain. juan.bueren@ciemat.es.
Trabectedin (Yondelis; ET-743) is a potent anticancer drug that binds to DNA by forming a covalent bond with a guanine in one strand and one or more hydrogen bonds with the opposite strand. Using a fluorescence-based melting assay, we show that one single trabectedin-DNA adduct increases the thermal stability of the double helix by >20 degrees C. As deduced from the analysis of phosphorylated H2AX and Rad51 foci, we observed that clinically relevant doses of trabectedin induce the formation of DNA double-strand breaks in human cells and activate homologous recombination repair in a manner similar to that evoked by the DNA interstrand cross-linking agent mitomycin C (MMC). Because one important characteristic of this drug is its marked cytotoxicity on cells lacking a functional Fanconi anemia (FA) pathway, we compared the response of different subtypes of FA cells to MMC and trabectedin. Our data clearly show that human cells with mutations in FANCA, FANCC, FANCF, FANCG, or FANCD1 genes are highly sensitive to both MMC and trabectedin. However, in marked contrast to MMC, trabectedin does not induce any significant accumulation of FA cells in G2-M. The critical relevance of FA proteins in the response of human cells to trabectedin reported herein, together with observations showing the role of the FA pathway in cancer suppression, strongly suggest that screening for mutations in FA genes may facilitate the identification of tumors displaying enhanced sensitivity to this novel anticancer drug.
PMID: 18483318
Structure, physiological role, and specific inhibitors of human thymidine kinase 2 (TK2): Present and future.
Pérez-Pérez MJ, Priego EM, Hernández AI, Familiar O, Camarasa MJ, Negri A, Gago F, Balzarini J.
Instituto de Química Médica (CSIC), E-28006 Madrid, Spain.
Human mitochondrial thymidine kinase (TK2) is a pyrimidine deoxynucleoside kinase (dNK) that catalyzes the phosphorylation of pyrimidine deoxynucleosides to their corresponding deoxynucleoside 5'-monophosphates by γ-phosphoryl transfer from ATP. In resting cells, TK2 is suggested to play a key role in the mitochondrial salvage pathway to provide pyrimidine nucleotides for mitochondrial DNA (mtDNA) synthesis and maintenance. However, recently the physiological role of TK2 turned out to have direct clinical relevance as well. Point mutations in the gene encoding TK2 have been correlated to mtDNA disorders in a heterogeneous group of patients suffering from the so-called mtDNA depletion syndrome (MDS). TK2 activity could also be involved in mitochondrial toxicity associated to prolonged treatment with antiviral nucleoside analogues like AZT and FIAU. Therefore, TK2 inhibitors can be considered as valuable tools to unravel the role of TK2 in the maintenance and homeostasis of mitochondrial nucleotide pools and mtDNA, and to clarify the contribution of TK2 activity to mitochondrial toxicity of certain antivirals. Highly selective TK-2 inhibitors having an acyclic nucleoside structure and efficiently discriminating between TK-2 and the closely related TK-1 have already been reported. It is actually unclear whether these agents efficiently reach the inner mitochondrial compartment. In the present review article, structural features of TK2, MDS-related mutations observed in TK2 and their role in MDS will be discussed. Also, an update on novel and selective TK2 inhibitors will be provided. c © 2008 Wiley Periodicals, Inc. Med Res Rev.
PMID: 18459168
Exploring Acyclic Nucleoside Analogues as Inhibitors of Mycobacterium tuberculosis Thymidylate Kinase.
Familiar O, Munier-Lehmann H, Negri A, Gago F, Douguet D, Rigouts L, Hernández AI, Camarasa MJ, Pérez-Pérez MJ.
Instituto de Química Médica (CSIC), Juan de la Cierva 3, 28006 Madrid, Spain,
Fax: (+34)91-564-4853.
In the search for novel inhibitors of the enzyme thymidine monophosphate kinase of Mycobacterium tuberculosis (TMPKmt), an attractive target for novel antituberculosis agents, we report herein the discovery of the first acyclic nucleoside analogues that potently and selectively inhibit TMPKmt. The most potent compounds in this series are (Z)-butenylthymines carrying a naphtholactam or naphthosultam moiety at position 4, which display Ki values of 0.42 and 0.27 µM, respectively. Docking studies followed by molecular dynamics simulations performed to rationalize the interaction of this new family of inhibitors with the target enzyme revealed a key interaction between the distal substituent and Arg 95 in the target enzyme. The fact that these inhibitors are more easily synthesizable than previously identified TMPKmt inhibitors, together with their potency against the target enzyme, makes them attractive lead compounds for further optimization.
PMID: 18418833
7,5'-O-dibenzylinosines: synthesis and studies on their conformational properties.
Casanova E, Aguado L, Jimeno ML, Gago F, Camarasa MJ, Pérez-Pérez MJ.
Instituto de Química Médica (CSIC), Madrid, Spain.
Reaction of 2',3'-O-isopropylidene inosine with benzyl bromide (1 h, rt) led to the 1,5'-O-dibenzylderivative 4, but by increasing the reaction time or the temperature, compound 4 is further transformed into the 1,7,5'-O-tribenzylinosine derivative 5. Similarly, the 7-methyl-1,5'-O-dibenzylderivative 6 has been synthesized from 4. The 1H-NMR spectra of 5 and 6 showed peculiar chemical shifts for geminal protons (H5' and H5'' of the ribose, and the CH2 of the benzyl groups). Preliminary NMR studies have been performed, including NOESY experiments that point toward the predominant existence of conformers that are stabilized by an electrostatic interaction between the positively charged imidazole of the base moiety and the high electron density of the 5'-benzyl substituent.
PMID: 18066882
Overcoming the Inadequacies or Limitations of Experimental Structures as Drug Targets by Using Computational Modeling Tools and Molecular Dynamics Simulations.
Marco E, Gago F.
Bioinformatics Unit, Centro de Biología Molecular "Severo Ochoa", Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain, Fax: (+34)914 974 799.
X-ray crystallography, NMR spectroscopy, and cryoelectron microscopy stand out as powerful tools that enable us to obtain atomic detail about biomolecules that can be potentially targeted by drugs. This knowledge is essential if virtual screening or structure-based ligand-design methods are going to be used in drug discovery. However, the macromolecule of interest is not always amenable to these types of experiment or, as is often the case, the conformation found experimentally cannot be used directly for docking studies because of significant changes between apo and bound forms. Furthermore, sometimes the desired insight into the binding mechanism cannot be gained because the structure of the ligand-receptor complex, not having been time-resolved, represents the endpoint of the binding process and therefore retains little or no information about the intermediate stages that led to its creation. Molecular dynamics (MD) simulations are routinely applied these days to the study of biomolecular systems with the aims of sampling configuration space more efficiently and getting a better understanding of the factors that determine structural stability and relevant biophysical and biochemical processes such as protein folding, ligand binding, and enzymatic reactions. This field has matured significantly in recent years, and strategies have been devised (for example, activated, steered, or targeted MD) that allow the calculated trajectories to be biased in attempts to properly shape a ligand binding pocket or simulate large-scale motions involving one or more protein domains. On the other hand, low-frequency motions can be simulated quite inexpensively by calculation of normal modes which allow the investigation of alternative receptor conformations. Selected examples in which these methods have been applied to several medicinal chemistry and in silico pharmacology endeavors are presented.
PMID: 17806089
Antitumor Activity, X-ray Crystal Structure, and DNA Binding Properties of Thiocoraline A, a Natural Bisintercalating Thiodepsipeptide
Negri A, Marco E, García-Hernández V, Domingo A, Llamas-Saiz AL, Porto-Sandá S, Riguera R, Laine W, David-Cordonnier MH, Bailly C, García-Fernández LF, Vaquero JJ, Gago F.
Departamento de Farmacología, Departamento de Bioquímica y Biología Molecular, and Departamento de Química Orgánica, Universidad de Alcalá, E-28871 Madrid, Spain, Unidad de Rayos X, Laboratorio Integral de Dinámica y Estructura de Biomoléculas José R. Carracido, Edificio CACTUS, Campus Sur, Universidad de Santiago de Compostela, 15782 Santiago de Compostela, Spain, Departamento de Química Orgánica, Facultad de Química, Universidad de Santiago de Compostela, E-15706 Santiago de Compostela, Spain, INSERM U-524 and Laboratoire de Pharmacologie Antitumorale du Centre Oscar Lambret, IRCL, Place de Verdun, F-59045 Lille, France, and PharmaMar S.A., Avda. de los Reyes 1, Polígono Industrial La Mina-Norte, E-28770 Colmenar Viejo, Madrid, Spain.
The marine natural product thiocoraline A displayed approximately equal cytotoxic activity at nanomolar concentrations in a panel of 12 human cancer cell lines. X-ray diffraction analyses of orthorhombic crystals of this DNA-binding drug revealed arrays of docked pairs of staple-shaped molecules in which one pendent hydroxyquinoline chromophore from each cysteine-rich molecule appears intercalated between the two chromophores of a facing molecule. This arrangement is in contrast to the proposed mode of binding to DNA that shows the two drug chromophores clamping two stacked base pairs, in agreement with the nearest-neighbor exclusion principle. Proof of DNA sequence recognition was obtained from both classical DNase I footprinting experiments and determination of the melting temperatures of several custom-designed fluorescently labeled oligonucleotides. A rationale for the DNA-binding behavior was gained when models of thiocoraline clamping a central step embedded in several octanucleotides were built and studied by means of unrestrained molecular dynamics simulations in aqueous solution.
PMID: 17571868
Supporting Information
Stepwise dissection and visualization of the catalytic mechanism of haloalkane dehalogenase LinB using molecular dynamics simulations and computer graphics.
Negri A, Marco E, Damborsky J, Gago F.
Department of Pharmacology, University of Alcala, E-28871 Alcala de Henares,
Madrid, Spain.
The different steps of the dehalogenation reaction carried out by LinB on three different substrates have been characterized using a combination of quantum mechanical calculations and molecular dynamics simulations. This has allowed us to obtain information in atomic detail about each step of the reaction mechanism, that is, substrate entrance and achievement of the near-attack conformation, transition state stabilization within the active site, halide stabilization, water molecule activation and subsequent hydrolytic attack on the ester intermediate with formation of alcohol, and finally product release. Importantly, no bias or external forces were applied during the whole procedure so that both intermediates and products were completely free to sample configuration space in order to adapt to the plasticity of the active site and/or search for an exit. Differences in substrate reactivity were found to be correlated with the ease of adopting the near-attack conformation and two different exit pathways were found for product release that do not interfere with substrate entrance. Additional support for the different entry and exit pathways was independently obtained from an examination of the enzyme's normal modes.
PMID: 17451982
Supporting Information
N1-Substituted Thymine Derivatives as Mitochondrial Thymidine Kinase (TK-2) Inhibitors
Hernández AI, Familiar O, Negri A, Rodríguez-Barrios F, Gago F, Karlsson A, Camarasa MJ, Balzarini J, Pérez-Pérez MJ
Instituto de Química Médica (C.S.I.C.), Juan de la Cierva 3, E-28006 Madrid, Spain, Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, Karolinska Institute, S-141 86 Huddinge/Stockholm, Sweden, Rega Institute for Medical Research, Katholieke Universiteit Leuven, B-3000 Leuven, Belgium.
Novel N1-substituted thymine derivatives related to 1-[(Z)-4-(triphenylmethoxy)-2-butenyl]thymine have been synthesized and evaluated against thymidine kinase-2 (TK-2) and related nucleoside kinases [i.e., Drosophila melanogaster deoxynucleoside kinase (Dm-dNK) and herpes simplex virus type 1 thymidine kinase (HSV-1 TK)]. The thymine base has been tethered to a distal triphenylmethoxy moiety through a polymethylene chain (n = 3-8) or through a (2-ethoxy)ethyl spacer. Moreover, substitutions at position 4 of one of the phenyl rings of the triphenylmethoxy moiety have been performed. Compounds with a hexamethylene spacer (18, 26b, 31) displayed the highest inhibitory values against TK-2 (IC50 = 0.3-0.5 µM). Compound 26b competitively inhibited TK-2 with respect to thymidine and uncompetitively with respect to ATP. A rationale for the biological data was provided by docking some representative inhibitors into a homology-based model of human TK-2. Moreover, two of the most potent TK-2 inhibitors (18 and 26b) that also inhibit HSV-1 TK were able to reverse the cytostatic activity of 1-(β-D-arabinofuranosyl)thymine (Ara-T) and ganciclovir in HSV-1 TK-expressing OST-TK(-)/HSV-1 TK(+) cell cultures.
PMID: 17181158
Supporting Information
Further Insight into the DNA Recognition Mechanism of Trabectedin from the Differential Affinity of Its Demethylated Analogue Ecteinascidin ET729 for the Triplet DNA Binding Site CGA
Marco E, David-Cordonnier MH, Bailly C, Cuevas C, Gago F
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, INSERM U-524 and Laboratoire de Pharmacologie Antitumorale du Centre Oscar Lambret, IRCL, Place de Verdun, F-59045 Lille, France, and PharmaMar S. A., Avda. de los Reyes, 1 Polígono Industrial La Mina, E-28770 Colmenar Viejo, Madrid, Spain.
Trabectedin and its N12-demethylated analogue ET729 bind covalently to the central guanine of selected DNA triplets. Although both drugs equally target several sites, including AGA, we show that covalent modification of CGA is only achieved by ET729. By means of molecular dynamics simulations of the precovalent complexes, we explain in atomic detail how such a simple structural modification brings about this notable change in the DNA-binding selectivity profiles of these two drugs.
PMID: 17154523
Supporting Information
Suppression of the acuH13 and acuH31 nonsense mutations in the carnitine/acylcarnitine translocase (acuH) gene of Aspergillus nidulans by the G265S substitution in the domain 2 of the release factor eRF1.
Martínez O, Marco E, Gago F, Laborda F, Ramón De Lucas J.
Departamento de Microbiología y Parasitología, Facultad de Farmacia, Campus Universitario, Universidad de Alcalá, Carretera Madrid-Barcelona Km 33, Alcalá de Henares ES-28871, Madrid, Spain.
A search for suppressors of the carnitine/acylcarnitine translocase (CACT) deficiency in Aspergillus nidulans permitted the identification of the suaE7 mutation, mapping at a new translational suppressor (suaE) gene. The suaE gene is essential in A. nidulans and encodes the eukaryotic release factor 1 (eRF1). The suaE7 mutation suppresses two acuH alleles (acuH13 and acuH31), both carrying nonsense mutations in the CACT encoding gene that involve the replacement of a CAG (Gln) codon with a premature TAG stop codon. In contrast, the suaE7 gene does not suppress the acuH20 amber nonsense mutation involving a TGG→TAG change. The phenotype associated to the suaE7 mutation strictly resembles that of mutants at the suaA and suaC genes, two translational suppressor genes previously identified, suggesting that their gene products might functionally interact in translation termination. Sequencing of the suaE7 gene allowed the identification of a mutation in the domain 2 of the omnipotent class-1 eukaryotic release factor involving the Gly265Ser substitution in the A. nidulans eRF1. This mutation creates a structural context unfavourable for normal eRF binding that allows the misreading of stop codons by natural suppressor tRNAs, such as the tRNAsGln. Structural analysis using molecular modelling of A. nidulans eRF1 domain 2 bearing the G265S substitution and computer simulation results suggest that this mutation might impair the necessary conformational changes in the eRF1 to optimally recognize the stop codon and simultaneously interact with the peptidyl transferase centre of the 60S ribosomal subunit.
PMID: 16971148
Mutational Pathways, Resistance Profile, and Side Effects of Cyanovirin Relative to Human Immunodeficiency Virus Type 1 Strains with N-Glycan Deletions in Their gp120 Envelopes.
Balzarini J, Van Laethem K, Peumans WJ, Van Damme EJ, Bolmstedt A, Gago F, Schols D.
Rega Institute for Medical Research, K.U. Leuven, Minderbroedersstraat 10, B-3000 Leuven, Belgium. jan.balzarini@rega.kuleuven.be.
Limited data are available on the genotypic and phenotypic resistance profile of the alpha-(1-2)mannose oligomer-specific prokaryotic lectin cyanovirin (CV-N). Therefore, a more systematic investigation was carried out to obtain a better view of the interaction between CV-N and human immunodeficiency virus type 1 (HIV-1) gp120. When HIV-1-infected CEM cell cultures were exposed to CV-N in a dose-escalating manner, a total of eight different amino acid mutations exclusively located at N-glycosylation sites in the envelope surface gp120 were observed. Six of the eight mutations resulted in the deletion of high-mannose type N-glycans (i.e., at amino acid positions 230, 332, 339, 386, 392, and 448). Two mutations (i.e., at position 136 and 160) deleted a complex type N-glycan in the variable V1/V2 domain of gp120. The level of phenotypic resistance of the mutated virus strains against CV-N generally correlated with the number of glycan deletions in gp120, although deletion of the glycans at N-230, N-392, and N-448 generally afforded a more pronounced CV-N resistance than other N-glycan deletions. However, the extent of the decrease of antiviral activity of CV-N against the mutated virus strains was markedly less pronounced than observed for alpha(1-3)- and alpha(1-6)-mannose-specific plant lectins Hippeastrum hybrid agglutinin (HHA) and Galanthus nivalis agglutinin (GNA), which points to the existence of a higher genetic barrier for CV-N. This is in agreement with a more consistent suppression of a wider variety of HIV-1 clades by CV-N than by HHA and GNA. Whereas the antiviral and in vitro antiproliferative activity of CV-N can be efficiently reversed by mannan, the pronounced mitogenic activity of CV-N on peripheral blood mononuclear cells was unaffected by mannan, indicating that some of the observed side effects of CV-N are unrelated to its carbohydrate specificity/activity.
PMID: 16912292
Cross-Talk between Nucleotide Excision and Homologous Recombination DNA Repair Pathways in the Mechanism of Action of Antitumor Trabectedin.
Herrero AB, Martin-Castellanos C, Marco E, Gago F, Moreno S.
Instituto de Biología Molecular y Celular del Cáncer, Consejo Superior de Investigaciones Científicas/Universidad de Salamanca, Campus Miguel de Unamuno, Salamanca and Departamento de Farmacología, Universidad de Alcalá, Alcalá de Henares, Madrid, Spain.
Trabectedin (Yondelis) is a potent antitumor drug that has the unique characteristic of killing cells by poisoning the DNA nucleotide excision repair (NER) machinery. The basis for the NER-dependent toxicity has not yet been elucidated but it has been proposed as the major determinant for the drug's cytotoxicity. To study the in vivo mode of action of trabectedin and to explore the role of NER in its cytotoxicity, we used the fission yeast Schizosaccharomyces pombe as a model system. Treatment of S. pombe wild-type cells with trabectedin led to cell cycle delay and activation of the DNA damage checkpoint, indicating that the drug causes DNA damage in vivo. DNA damage induced by the drug is mostly caused by the NER protein, Rad13 (the fission yeast orthologue to human XPG), and is mainly repaired by homologous recombination. By constructing different rad13 mutants, we show that the DNA damage induced by trabectedin depends on a 46-amino acid region of Rad13 that is homologous to a DNA-binding region of human nuclease FEN-1. More specifically, an arginine residue in Rad13 (Arg961), conserved in FEN1 (Arg314), was found to be crucial for the drug's cytotoxicity. These results lead us to propose a model for the action of trabectedin in eukaryotic cells in which the formation of a Rad13/DNA-trabectedin ternary complex, stabilized by Arg961, results in cell death.
PMID: 16912194
[Supplementary Data]
Dimerization inhibitors of HIV-1 reverse transcriptase, protease and integrase: A single mode of inhibition for the three HIV enzymes?
Camarasa MJ, Velázquez S, San-Félix A, Pérez-Pérez MJ, Gago F.
Instituto de Química Médica (C.S.I.C.), Juan de la Cierva 3, E-28006 Madrid, Spain
The genome of human immunodeficiency virus type 1 (HIV-1) encodes 15 distinct proteins, three of which provide essential enzymatic functions: a reverse transcriptase (RT), an integrase (IN), and a protease (PR). Since these enzymes are all homodimers, pseudohomodimers or multimers, disruption of protein-protein interactions in these retroviral enzymes may constitute an alternative way to achieve HIV-1 inhibition. A growing number of dimerization inhibitors for these enzymes is being reported. This mini review summarizes some approaches that have been followed for the development of compounds that inhibit those three enzymes by interfering with the dimerization interfaces between the enzyme subunits.
PMID: 16872687
Involvement of novel human immunodeficiency virus type 1 reverse transcriptase mutations in the regulation of resistance to nucleoside inhibitors.
Svicher V, Sing T, Santoro MM, Forbici F, Rodriguez-Barrios F, Bertoli A, Beerenwinkel N, Bellocchi MC, Gago F, d'Arminio Monforte A, Antinori A, Lengauer T, Ceccherini-Silberstein F, Perno CF.
Department of Experimental Medicine, University of Rome Tor Vergata, Via
Montpellier 1, 00133 Rome, Italy. ceccherini@med.uniroma2.it.
We characterized 16 additional mutations in human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) whose role in drug resistance is still unknown by analyzing 1,906 plasma-derived HIV-1 subtype B pol sequences from 551 drug-naive patients and 1,355 nucleoside RT inhibitor (NRTI)-treated patients. Twelve mutations positively associated with NRTI treatment strongly correlated both in pairs and in clusters with known NRTI resistance mutations on divergent evolutionary pathways. In particular, T39A, K43E/Q, K122E, E203K, and H208Y clustered with the nucleoside analogue mutation 1 cluster (NAM1; M41L+L210W+T215Y). Their copresence in this cluster was associated with an increase in thymidine analogue resistance. Moreover, treatment failure in the presence of K43E, K122E, or H208Y was significantly associated with higher viremia and lower CD4 cell count. Differently, D218E clustered with the NAM2 pathway (D67N+K70R+K219Q+T215F), and its presence in this cluster determined an increase in zidovudine resistance. In contrast, three mutations (V35I, I50V, and R83K) negatively associated with NRTI treatment showed negative correlations with NRTI resistance mutations and were associated with increased susceptibility to specific NRTIs. In particular, I50V negatively correlated with the lamivudine-selected mutation M184V and was associated with a decrease in M184V/lamivudine resistance, whereas R83K negatively correlated with both NAM1 and NAM2 clusters and was associated with a decrease in thymidine analogue resistance. Finally, the association pattern of the F214L polymorphism revealed its propensity for the NAM2 pathway and its strong negative association with the NAM1 pathway. Our study provides evidence of novel RT mutational patterns that regulate positively and/or negatively NRTI resistance and strongly suggests that other mutations beyond those currently known to confer resistance should be considered for improved prediction of clinical response to antiretroviral drugs.
PMID: 16809324
Discovery of TSAO derivatives with an unusual HIV-1 activity/resistance profile.
de Castro S, García-Aparicio C, Van Laethem K, Gago F, Lobatón E, De Clercq E, Balzarini J, Camarasa MJ, Velázquez S.
Instituto de Química Médica (C.S.I.C.), Juan de la Cierva 3, E-28006 Madrid, Spain
The very first TSAO derivative that lacks the 4''-amino group at the 3'-spiro moiety (compound 3) has been prepared and the effect of this modification on the activity/resistance profile has been evaluated. This molecule proved HIV-1 specific NNRTI-characteristic). A mixture of wild-type and V106V/A or L234L/I mutations were found in the RT of some, but not all compound 3-resistant virus strains. Compound 3 does not select for the TSAO-specific E138K mutation in the RT. However, the compound markedly lost its antiviral potential against a variety of virus strains that contain NNRTI-characteristic mutations in RT including E138K. The deaminated TSAO compound must fit differently in the HIV-1 RT enzyme than its prototype TSAO-m3T.
PMID: 16616962
Mitochondrial Thymidine Kinase Inhibitors.
Pérez-Pérez MJ, Hernández AI, Priego EM, Rodríguez-Barrios F, Gago F, Camarasa MJ, Balzarini J.
Instituto de Química Médica (CSIC), Juan de la Cierva 3, E-28006 Madrid, Spain. mjperez@iqm.csic.es.
Mitochondrial thymidine kinase or TK-2 belongs to the family of mammalian deoxynucleoside kinases (dNKs) that catalyze the phosphorylation of deoxynucleosides to their corresponding deoxynucleoside monophosphates by γ-phosphoryl transfer of ATP. These enzymes are instrumental in the activation of deoxynucleoside analogues with biological and therapeutic properties. Moreover, dNKs are fundamental to maintain dNTPs pools for DNA synthesis and repair. TK-2 has a mitochondrial localization and is the only thymidine kinase that is physiologically active in non-proliferating and resting cells. Several recent investigations point to an important role of TK-2 in the maintenance of mitochondrial dNTPs pools. Indeed, mutations in the gene encoding TK-2 have been associated with mitochondrial DNA (mtDNA) depletion that mostly affects skeletal muscle. Moreover, TK-2 has been suggested to be implicated in mitochondrial toxicity associated to prolonged treatments with nucleoside analogues (i.e AZT for the treatment of AIDS patients). In this scenario, TK-2 inhibitors could be a useful tool to further clarify both the physiological role of TK-2 in the maintenance of mitochondrial dNTP pools, and the possible contribution of TK-2 to the mitochondrial toxicity of pyrimidine nucleoside analogues. In the present article we review the most recent literature covering different aspects of TK-2 as well as published TK-2 inhibitors, with special emphasis on acyclic nucleoside analogues that have been described by our research groups and whose prototype compound is 1-[(Z)-4-(triphenylmethoxy)-2-butenyl]thymine.
PMID: 16305527
Role of stacking interactions in the binding sequence preferences of DNA bis-intercalators: insight from thermodynamic integration free energy simulations.
Marco E, Negri A, Luque FJ, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
The major structural determinant of the preference to bind to CpG binding sites on DNA exhibited by the natural quinoxaline bis-intercalators echinomycin and triostin A, or the quinoline echinomycin derivative, 2QN, is the 2-amino group of guanine (G). However, relocation of this group by means of introduction into the DNA molecule of the 2-aminoadenine (=2,6-diaminopurine, D) base in place of adenine (A) has been shown to lead to a drastic redistribution of binding sites, together with ultratight binding of 2QN to the sequence DTDT. Also, the demethylated triostin analogs, TANDEM and CysMeTANDEM, which bind with high affinity to TpA steps in natural DNA, bind much less tightly to CpI steps, despite the fact that both adenosine and the hypoxanthine-containing nucleoside, inosine (I), provide the same hydrogen bonding possibilities in the minor groove. To study both the increased binding affinity of 2QN for DTDT relative to GCGC sites and the remarkable loss of binding energy between CysMeTANDEM and ICIC compared with ATAT, a series of thermodynamic integration free energy simulations involving conversions between DNA base pairs have been performed. Our results demonstrate that the electrostatic component of the stacking interactions between the heteroaromatic rings of these compounds and the bases that make up the intercalation sites plays a very important role in the modulation of their binding affinities.
PMID: 16282585 10.1093/nar/gki916
[Supplementary data]
Binding of 5'-GMP to the GluR2 AMPA Receptor: Insight from Targeted Molecular Dynamics Simulations.
Mendieta J, Gago F, Ramirez G.
Centro de Biología Molecular Severo Ochoa, Universidad Autónoma,
E-28049 Cantoblanco, Madrid, Spain, and Departamento de Farmacología,
Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid,
Spain.
Guanine nucleotides behave as competitive antagonists at ionotropic glutamate receptors and show neuroprotective activity in different experimental excitotoxicity paradigms, both in vivo and in cultured cell preparations. Taking 5'-GMP as the reference nucleotide, we have tried to understand how these molecules interact with the agonist-binding site of the GluR2 α-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptor. Using a crystallographic model of the ligand-binding core of the GluR2 receptor in complex with kainate, we have previously analyzed the structural changes associated to the binding of agonists to the receptor and suggested a mechanism for the coupling of agonist binding to channel gating. In the present investigation we used the structure of the apo form of the receptor to probe the primary interactions between GMP and GluR2 by means of an automated docking program. A targeted molecular dynamics (TMD) simulation procedure was subsequently used to force the closing of the protein and to study the rearrangement of the ligand and surrounding amino acids. The resulting structure provides a plausible model of the nucleotide-receptor complex. Indirect support for the validity of our approach was obtained when the same methodology was shown to yield structures of the kainate-GluR2 and 6,7-dinitroquinoxaline-2,3-dione (DNQX)-GluR2 complexes that were in very good agreement with the published crystallographic structures. Both the stacking interaction between the phenyl ring of Tyr73 and the purine ring of GMP and a salt bridge between the phosphate group of GMP and Arg108 in the S1 domain, together with several hydrogen bonds, are proposed to secure the anchoring of GMP to the agonist-binding site. Unlike conventional competitive antagonists, such as DNQX, occupancy of the site by GMP still allows receptor segments S1 and S2 to close tightly around GMP without interacting with the critical residue Glu209 that triggers channel opening. Thus, GMP appears to be rather a false agonist than a competitive antagonist. This fact and the nature of the energy barriers that stabilize GMP bound to the closed form of the receptor provide an explanation for the unusual behavior of some guanine nucleotides in ligand-displacement experiments.
PMID: 16262247
Improving the Antiviral Efficacy and Selectivity of HIV-1 Reverse Transcriptase Inhibitor TSAO-T by the Introduction of Functional Groups at the N-3 Position.
Bonache MC, Chamorro C, Velázquez S, De Clercq E, Balzarini J, Barrios FR, Gago F, Camarasa MJ, San-Félix A.
Instituto de Química Médica (C.S.I.C.), Madrid, Spain, Rega Institute for Medical Research, K. U. Leuven, Leuven, Belgium, and Departamento de Farmacología, Universidad de Alcalá, Alcalá de Henares, Madrid, Spain.
Novel derivatives of the anti-HIV-1 agent, TSAO-T, bearing at the N-3 position a variety of polar, lipophilic, or aromatic groups linked to that position through flexible polymethylene linkers of different length, were prepared and evaluated for their anti-HIV activity. Several compounds (within the series of polar bearing groups) exhibited a 2-6-fold improved antiviral potency with regard to the lead compound, TSAO-T. Moreover, some of the most active N-3 TSAO derivatives retain antiviral activity against the TSAO-T-resistant HIV-1 strain (Glu138 → Lys). Interestingly, the N-methylcarboxamide derivative 17 was 5- to 6-fold more active (IC50): 0.56 µM) against recombinant HIV-1 reverse transcriptase than the lead compound, thus becoming the most active TSAO derivative synthesized so far. On the other hand, the N-3 methylcarboxamide TSAO derivative 12 turned out to have the highest selectivity index yet reported for this class of compounds (around >/=12 000).
PMID: 16220981
[Supporting Information]
DNA structural similarity in the 2:1 complexes of the antitumor drugs Yondelis (trabectedin) and chromomycin A3 with an oligonucleotide sequence containing two adjacent TGG binding sites on opposing strands.
Marco E, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain. federico.gago@uah.es.
Yondelis (trabectedin) is an antitumor ecteinascidin that binds covalently to the 2-amino group of the central guanine in the minor groove of selected DNA pyrimidine-G-G and purine-G-C triplets. Chromomycin A3 is an aureolic acid derivative that binds non-covalently to the DNA minor groove in G/C-rich triplet sites as a metal-chelated dimer. Despite their different binding modes, the cytotoxicity profiles of these two drugs, as assessed in the COMPARE analysis carried out by the National Cancer Institute on data from 60 human tumor cell lines, are highly correlated (Pearson correlation coefficient of 0.96). We now report that in a oligonucleotide containing the "natural bending element" TGGCCA, the structural distortions inflicted by the tail-to-tail bonding of two trabectedin molecules to adjacent target sites on opposing strands are strikingly similar to those observed in a crystal containing d(TTGGCCAA2 and two bound chromomycin A3 molecules arranged in a head-to-tail orientation in the minor groove. In both complexes the double helix is characterized by being considerably unwound and possessing a notably widened minor groove. Binding of the drugs to this sequence could be favored by the distinct bends at each of the TpG steps that are already present in the free oligoucleotide. Simultaneous drug binding to the two strands in the manner described here is proposed to stabilize the helical structure of duplex DNA so as to prevent or hamper strand separation and stall replication and transcription forks.
PMID: 16150929
| |
|
2B0I Computer model of a covalent complex between d(GTATGGCCATAC) and two molecules of antitumor drug trabectedin |
High Sequence Conservation of Human Immunodeficiency Virus Type 1 Reverse Transcriptase under Drug Pressure despite the Continuous Appearance of Mutations.
Ceccherini-Silberstein F, Gago F, Santoro M, Gori C, Svicher V, Rodríguez-Barrios F, d'Arrigo R, Ciccozzi M, Bertoli A, Monforte A, Balzarini J, Antinori A, Perno CF.
Department of Experimental Medicine, University of Rome Tor Vergata, Via
Montpellier 1, 00133 Rome, Italy. ceccherini@med.uniroma2.it.
To define the extent of sequence conservation in human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) in vivo, the first 320 amino acids of RT obtained from 2,236 plasma-derived samples from a well-defined cohort of 1,704 HIV-1-infected individuals (457 drug naive and 1,247 drug treated) were analyzed and examined in structural terms. In naive patients, 233 out of these 320 residues (73%) were conserved (<1% variability). The majority of invariant amino acids clustered into defined regions comprising between 5 and 29 consecutive residues. Of the nine longest invariant regions identified, some contained residues and domains critical for enzyme stability and function. In patients treated with RT inhibitors, despite profound drug pressure and the appearance of mutations primarily associated with resistance, 202 amino acids (63%) remained highly conserved and appeared mostly distributed in regions of variable length. This finding suggests that participation of consecutive residues in structural domains is strictly required for cooperative functions and sustainability of HIV-1 RT activity. Besides confirming the conservation of amino acids that are already known to be important for catalytic activity, stability of the heterodimer interface, and/or primer/template binding, the other 62 new invariable residues are now identified and mapped onto the three-dimensional structure of the enzyme. This new knowledge could be of help in the structure-based design of novel resistance-evading drugs.
PMID: 16051864
Role of T139 in the human immunodeficiency virus type 1 (HIV-1) reverse transcriptase (RT) sensitivity to (+)-calanolide A.
Auwerx J, Rodríguez-Barrios F, Ceccherini-Silberstein F, San-Felix A, Velázquez S, De Clercq E, Camarasa MJ, Perno CF, Gago F, Balzarini J.
Rega Institute for Medical Research, Minderbroedersstraat 10, B-3000 Leuven, Belgium.
The coumarins represent a unique class of non-nucleoside reverse transcriptase inhibitors (NNRTIs) that were isolated from tropical plants. (+)-Calanolide A, the most potent compound of this class, selects for the T139I resistance mutation in HIV-1 reverse transcriptase (RT). Seven RTs mutated at amino acid position 139 (A, K, Y, D, I, S and Q) were constructed by site-directed mutagenesis. The mutant T139Q enzyme retained full catalytic activity compared to wild-type RT, while the mutant T139I, T139S and T139A RTs retained only 85% to 50% of the activity. Mutant T139K, T139D and T139Y RTs had seriously impaired catalytic activities. The mutations in the T139I and T139D RTs were shown to destabilize the RT heterodimer. (+) Calanolide A lost inhibitory activity (up to 20-fold) against the mutant T139Y, T139Q, T139K and T139I enzymes. All mutant enzymes retained marked susceptibility towards the other NNRTIs including nevirapine, delavirdine, efavirenz, the thiocarboxanilide UC-781, the quinoxaline GW867420X and the TSAO derivatives, and the nucleotide inhibitor ddGTP. The fact that the T139I RT (i) proved resistant to (+)-calanolide A, (ii) represents a catalytically efficient enzyme, and (iii) requires only a single transition point mutation (ACA→ATA) in codon 139 appears to explain why mutant T139I RT virus strains, but not virus strains containing other amino acid changes at this position, predominantly emerge in cell cultures under (+)-calanolide A pressure.
PMID: 15961674
Molecular Determinants of Topoisomerase I Poisoning by Lamellarins: Comparison with Camptothecin and Structure-Activity Relationships.
Marco E, Laine W, Tardy C, Lansiaux A, Iwao M, Ishibashi F, Bailly C, Gago F
Departamento de Farmacología, Universidad de Alcalá, E-28871 Madrid, Spain, INSERM U-524 and Laboratoire de Pharmacologie Antitumorale du Centre Oscar Lambret, IRCL, Place de Verdun, F-59045 Lille, France, and Faculty of Engineering and Faculty of Fisheries, Nagasaki University, Nagasaki 852-8521, Japan
A series of lamellarin derivatives have been studied as topoisomerase I (Top1) inhibitors. Molecular models of the ternary complexes formed between the DNA-Top1 ensemble and lamellarin D (LMD) or camptothecin (CPT) fully intercalated into the duplex DNA have been built and studied by means of nanosecond molecular dynamics simulations in aqueous solution. Our results show that the 20-OH and 8-OH of LMD can participate in hydrogen-bonding interactions with the side chains of Glu356 and Asn722, respectively, the latter being consistent with the finding that CEM/C2 cells, which are resistant to CPT, are cross-resistant to LMD. Our models also account for the observation that LMD stabilizes Top1 cleavage at CG sites in addition to the TG sites observed for CPT and rationalize the structure-activity relationships within the series. The deleterious effect of replacing the 20-OH in LMD with a hydrogen was confirmed using a set of thermodynamic integration free energy simulations.
PMID: 15916431
[Supporting
Information]
The Molecular Basis of Resilience to the Effect of the Lys103Asn Mutation in Non-Nucleoside HIV-1 Reverse Transcriptase Inhibitors Studied by Targeted Molecular Dynamics Simulations
Rodríguez-Barrios F, Balzarini J, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, and Rega Institute for Medical
Research, K. U. Leuven, B-3000 Leuven, Belgium.
A series of targeted molecular dynamics simulations have been carried out in an attempt to assess the effect that the common Lys103Asn mutation in HIV-1 reverse transcriptase (RT) has on the binding of three representative non-nucleoside RT inhibitors (NNRTI), nevirapine, efavirenz, and etravirine. We have shown previously that, in the absence of an incoming inhibitor, creation of the NNRTI binding pocket is hampered due to the existence of a hydrogen bond between the side chains of Asn103 and Tyr188 for which no equivalent exists in the wild-type enzyme. As an extension of this work, we now apply the same methodology to drive the enzyme's conformation from the unbound state to the drug-bound state in the presence of the NNRTI. The location of each drug outside the binding pocket was determined by an automated docking program, and steering into the binding pocket followed a route that is likely to represent the actual entrance pathway. The additional hurdle to inhibitor entry imposed by the extra Asn103-Tyr188 hydrogen bond is seen to affect each NNRTI differently, with the ability to disrupt this interaction increasing in the order etravirine >> efavirenz > nevirapine, in good accord with the experimental findings. This coherent picture strongly suggests that attempts to overcome resistance through structure-based drug design may be considerably more successful if dynamic structural aspects of the type studied here are considered, particularly in cases where binding energy-based structure-activity relationship methods are unable to provide the required information.
PMID: 15898808
[Supporting
Information]
The N137 and P140 amino acids in the p51 and the P95 amino acid in the p66 subunit of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase are instrumental to maintain catalytic activity and to design new classes
of anti-HIV-1 drugs
Auwerx J, Van Nieuwenhove J, Rodríguez-Barrios F, de Castro S, Velázquez S, Ceccherini-Silberstein F, De Clercq E, Camarasa MJ, Perno CF, Gago F, Balzarini J
Rega Institute for Medical Research, K.U. Leuven, B-3000 Leuven, Belgium, Department of Pharmacology, University of Alcalá, E-28871 Alcalá de Henares, Spain, Instituto de Química Médica, C.S.I.C., E-28006 Madrid, Spain, Department of Experimental Medicine, University of Rome Tor Vergata, I-00133 Rome, Italy.
Amino acids N137 and P140 in the p51 subunit of HIV-1 reverse transcriptase (RT) are part of the β7β8- that contributes to the formation of the base of the non-nucleoside RT inhibitor (NNRTI)-binding pocket and makes up a substantial part of the dimerization interface. Amino acid P95 in p66 also markedly contributes to the dimerization binding energy. Nine RT mutants at amino acid 137 were constructed bearing the mutations Y, K, T, D, A, Q, S, H or E. The prolines at amino acid positions 95 and 140 were replaced by alanine in separate enzymes. We found that all mutant RT enzymes showed a dramatically decreased RNA-dependent DNA polymerase activity. None of the mutant RT enzymes showed marked resistance against any of the clinically used NNRTIs but they surprisingly lost significant sensitivity for NRTIs such as ddGTP. The denaturation analyses of the mutant RTs by urea are suggestive for a relevant role of N137 in the stability of the RT heterodimer and support the view that the β7β8 loop in p51 is a hot spot for RT dimerization and instrumental for efficient polymerase catalytic activity. Consequently, N137 and P140 in p51 and P95 in p66 should be attractive targets in the design of new structural classes of RT inhibitors aimed at compromising the optimal interaction of the β7β8 loop in p51 at the p66/p51 dimerization interface.
PMID: 15848161
The Amino Acid Asn136 in HIV-1 Reverse Transcriptase (RT) Maintains Efficient Association of Both RT Subunits and Enables the Rational Design of Novel RT Inhibitors.
Balzarini J, Auxerx J, Rodriguez-Barrios F, Chedad A, Farkas V, Ceccherini-Silberstein F, Garcia-Aparicio C, Velazquez S, De Clercq E, Perno CF, Camarasa MJ, Gago F.
Rega Institute for Medical Research, Minderbroedersstraat 10, B-3000 Leuven, Belgium. jan.balzarini@rega.kuleuven.be.
The highly conserved N136 is in close vicinity of the non-nucleoside reverse transcriptase (RT) inhibitor (NNRTI)-specific lipophilic pocket
of human immunodeficiency virus type 1 (HIV-1) RT. Site-directed mutagenesis has revealed that the catalytic activity of HIV-1 RT mutated at position
N136 is heavily compromised. Only 0.07 to 2.1% of wild-type activity is retained depending on the nature of the amino acid change at position 136.
The detrimental effect of the mutations at position 136 occurred when the mutated amino acid was present in the p51 subunit, but not in the p66 subunit of the p51/p66 RT heterodimer. All mutant 136 enzymes could be inhibited by second-generation NNRTIs such as efavirenz. They were also markedly more sensitive to the inactivating (denaturing) effect of urea (and acetonitrile) than wild-type RT, and the degree of increased urea sensitivity was highly correlated with the degree of (lower) catalytic activity of the mutant enzymes. Replacing wild-type N136 in HIV-1 RT by other amino acids resulted in notably increased amounts of free p51 and p66 monomers in the RT preparation. Our findings identify a structural/functional role for N136 in stabilization of the RT p66/p51 dimer and provide hints for the rational design of novel NNRTIs or drugs targeting either N136 in the β7-β8 loop of p51 or its anchoring point on p66 (the peptide backbone of H96) so as to interfere with the RT dimerisation process and/or with the structural support that the p51 subunit provides to the p66 subunit and which is essential for catalytic activity of the enzyme.
PMID: 15833734
Quantitative Analysis of Substrate Specificity of Haloalkane Dehalogenase LinB from Sphingomonas paucimobilis UT26.
Kmunicek J, Hynkova K, Jedlicka T, Nagata Y, Negri A, Gago F, Wade RC, Damborsky J.
National Centre for Biomolecular Research, Masaryk University, Kotlarska 2, 61137 Brno, Czech Republic, Department of Environmental Life Sciences,
Graduate School of Life Sciences, Tohoku University, 2-1-1 Katahira, Sendai 980-8577, Japan, Department of Pharmacology, University of Alcala, E-28871
Alcalá de Henares, Madrid, Spain, and EML Research, Villa Bosch, Schloss-Wolfsbrunnenweg 33, D-69118 Heidelberg, Germany.
Haloalkane dehalogenases are microbial enzymes that cleave a carbon-halogen bond in halogenated compounds. The haloalkane dehalogenase LinB, isolated from Sphingomonas paucimobilis UT26, is a broad-specificity enzyme. Fifty-five halogenated aliphatic and cyclic hydrocarbons were tested for dehalogenation with the LinB enzyme. The compounds for testing were systematically selected using a statistical experimental design. Steady-state kinetic constants Km and kcat were determined for 25 substrates that showed detectable cleavage by the enzyme and low abiotic hydrolysis. Classical quantitative structure-activity relationships (QSARs) were used to correlate the kinetic constants with molecular descriptors and resulted in a model that explained 94% of the experimental data variability. The binding affinity of the tested substrates for this haloalkane dehalogenase correlated with hydrophobicity, molecular surface, dipole moment, and volume:surface ratio. Binding of the substrate molecules in the active site pocket of LinB depends nonlinearly on the size of the molecules. Binding affinity increases with increasing substrate size up to a chain length of six carbon atoms and then decreases. Comparative binding energy (COMBINE) analysis was then used to identify amino acid residues in LinB that modulate its substrate specificity. A model with three statistically significant principal components explained 95% of the experimental data variability. van der Waals interactions between substrate molecules and the enzyme dominated the COMBINE model, in agreement with the importance of substrate size in the classical QSAR model. Only a limited number of protein residues (6-8%) contribute significantly to the explanation of variability in binding affinities. The amino acid residues important for explaining variability in binding affinities are as follows: (i) first-shell residues Asn38, Asp108, Trp109, Glu132, Ile134, Phe143, Phe151, Phe169, Val173, Trp207, Pro208, Ile211, Leu248, and His272, (ii) tunnel residues Pro144, Asp147, Leu177, and Ala247, and (iii) second-shell residues Pro39 and Phe273. The tunnel and the second-shell residues represent the best targets for modulating specificity since their replacement does not lead to loss of functionality by disruption of the active site architecture. The mechanism of molecular adaptation toward a different specificity is discussed on the basis of quantitative comparison of models derived for two protein family members.
PMID: 15736949
Phosphorylation modulates the α-helical structure and polymerization of a peptide from the third tau microtubule-binding repeat.
Mendieta J, Fuertes MA, Kunjishapatham R, Santa-María I, Moreno FJ, Alonso C, Gago F, Muñoz V, Avila J, Hernández F.
Centro de Biología Molecular Severo Ochoa CSIC/UAM, Fac. Ciencias, Universidad Autónoma de Madrid, Cantoblanco, 28049 Madrid, Spain
Paired helical filaments (PHFs) isolated from patients with Alzheimers disease (AD) mainly consist of the microtubule-associated protein tau in
a hyperphosphorylated form. It has been found that PHFs are the first example of pathological protein aggregation associated with formation of α-helices [Biochemistry (2002) 41, 71505]. In an effort to investigate the interplay between phosphorylation and the putative role of short regions of α-helix in the polymerization of tau, we have focused on the region of tau encompassing residues 317 to 335. This region is able to form protein fibrils in vitro and has two serines that are often found phosphorylated in PHFs. Using trifluoroethanol as an indicator of the α-helix, we find that the stability of the α-helix conformation is enhanced by phosphorylation. Circular dichroism data show that the phosphorylated peptide in water presents a content in α-helix similar to the unphosphorylated peptide at 40% of trifluoroethanol. Phosphorylation also stimulates the effect of juglone in promoting the in vitro polymerization. Furthermore, Fourier transformed infrared spectroscopy of samples of phosphorylated peptide polymerized with juglone renders a spectrum with maxima at ~1665 and ~1675 cm-1,
which are suggestive of a mixture of turns and α-helix conformations. Our results provide a direct mechanistic connection between phosphorylation and polymerization in tau. The connection between phosphorylation and polymerization appears to involve formation of α-helix structure.
PMID: 15652175
Understanding ET-18-OCH3 (edelfosine): a selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor.
Mollinedo F, Gajate C, Martin-Santamaria S, Gago F
Centro de Investigación del Cáncer, Instituto de Biología Molecular y Celular del Cáncer, CSIC-Universidad de Salamanca, Campus Miguel de Unamuno, E-37007 Salamanca, Spain. fmollin@usal.es
Synthetic ether-linked analogues of phosphatidylcholine and lysophosphatidylcholine, collectively named as antitumour lipids (ATLs), were initially synthesized in the late 60s, but have attracted a renewed interest since the finding that the ether lipid 1-O-octadecyl-2-O-methyl-rac-glycero-3-phosphocholine (ET-18-OCH3, edelfosine), a synthetic analogue of 2-lysophosphatidylcholine
considered the ATL prototype, induces a selective apoptotic response in tumour cells, sparing normal cells. Unlike most chemotherapeutic agents
currently used, ET-18-OCH3 does not interact with DNA, but act at the cell membrane, and thereby its effects seem to be independent of
the proliferative state of target cells. Each part of the molecular structure of ET-18-OCH3 is important for its optimal proapoptotic activity. Recent progress has unveiled the molecular mechanism underlying the apoptotic action of ET-18-OCH3, involving membrane rafts and Fas/CD95 death receptor, and has led to the proposal of a two-step model for the ET-18-OCH3 selective action on cancer cells, namely: a) ET-18-OCH3 uptake into the tumour cell, but not in normal cells; b) intracellular activation of Fas/CD95 through its translocation and capping into membrane rafts. ET-18-OCH3 constitutes the first antitumour drug acting through the intracellular activation of the Fas/CD95 death receptor. Computational docking studies have allowed us to propose a molecular model for the putative interaction of ET-18-OCH3 with the intracellular Fas/CD95 death domain. This novel mechanism of action represents a new way to target tumour
cells in cancer chemotherapy and can be of interest as a new framework in designing novel and more selective proapoptotic antitumour drugs.
PMID: 15579006
Understanding the Basis of Resistance in the Irksome Lys103Asn HIV-1 Reverse Transcriptase Mutant Through Targeted Molecular Dynamics Simulations.
Rodríguez-Barrios F, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
Results of targeted molecular dynamics simulations confirm the existence of a higher energy barrier for creation of the pocket where non-nucleoside reverse transcriptase inhibitors bind in the K103N mutant enzyme relative to wild-type.
PMID: 15563158
[Supporting
Information]
Modelling and Simulation: A Computational Perspective in Anticancer Drug Discovery.
Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
The availability of high-quality molecular graphics tools in the public domain is changing the way macromolecular structure is perceived by researchers, educators and students alike. Computational methods have become increasingly important in a number of areas such as comparative or homology modelling, functional site location, characterization of ligand-binding sites in proteins, docking of small molecules into protein binding sites, protein-protein docking, and molecular dynamics simulations. The results obtained yield information that sometimes is beyond current experimental possibilities and can be used to guide and improve a vast array of experiments. On the basis of our improved level of understanding of molecular recognition and the widespread availability of target structures, it is reasonable to assume that computational methods will continue aiding not only in the design and interpretation of hypothesis-driven experiments in the field of cancer research but also in the rapid generation of new hypotheses.
PMID: 15379692
In silico activation of Src tyrosine kinase reveals the molecular basis for intramolecular autophosphorylation.
Mendieta J, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
Structural data suggest that important hinge-bending motions of the two lobes that shape the catalytic domain of Src tyrosine kinase, together
with reorganization of an α helix (helix C), are needed for the activation loop to adopt the catalytically competent conformation. The phosphorylation of a Tyr residue (Tyr-416) in this loop also seems to be essential for enzyme activation. However, no information is available about the dynamics of this activation process. By comparing the inactive and active forms of the catalytic domains of Src and Lck, another member of the Src family, we first identified a short stretch that can act as a hinge for the interlobe motion. The opening of the lobes was then simulated using a targeted molecular dynamics approach. The results obtained suggested that pulling the two lobes apart is not enough to induce the required conformational change in the activation loop. Rather unexpectedly, however, swinging of the lobes situated Tyr-416 in a suitable position for intramolecular autophosphorylation, and further simulation of Tyr-416-phosphorylated Src in the presence of ADP did then result in a conformational change that placed the activation loop in a position similar to that found in the active open conformation of Lck. Taken together, our results establish a physical link between intramolecular autophosphorylation and loop activation.
PMID: 15363460
[Supporting Information]
 |
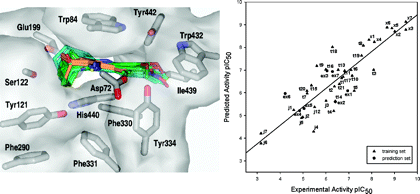 |
J Med Chem. 2004 Aug 26; 47(18) 4471-4482 |
Modulation of Binding Strength in Several Classes of Active Site Inhibitors of Acetylcholinesterase Studied by Comparative Binding Energy Analysis.
Martín-Santamaría S, Muñoz-Muriedas J, Luque FJ, Gago F.
Departament de Fisicoquímica, Facultat de Farmàcia, Universitat de Barcelona, Av. Diagonal 643, 08028 Barcelona, Spain, and Departamento de Farmacología, Universidad de Alcalá, 28871 Alcalá de Henares, Madrid, Spain.
The comparative binding energy (COMBINE) methodology has been used to identify the key residues that modulate the inhibitory potencies of three
structurally different classes of acetylcholinesterase inhibitors (tacrines, huprines, and dihydroquinazolines) targeting the catalytic active site
of this enzyme. The extended set of energy descriptors and the partial least-squares methodology used by COMBINE analysis on a unique training
set containing all the compounds yielded an interpretable model that was able to fit and predict the activities of the whole series of inhibitors
reasonably well (r2 = 0.91 and q2 = 0.76, 4 principal components). A more robust model (q2 = 0.81 and SDEP = 0.25,
3 principal components) was obtained when the same chemometric analysis was applied to the huprines set alone, but the method was unable to provide predictive models for the other two families when they were treated separately from the rest. This finding appears to indicate that the enrichment in chemical information brought about by the inclusion of different classes of compounds into a single training set can be beneficial when an internally consistent set of pharmacological data can be derived. The COMBINE model was externally validated when it was shown to predict the activity of an additional set of compounds that were not employed in model construction. Remarkably, the differences in inhibitory potency within the whole series were found to be finely tuned by the electrostatic contribution to the desolvation of the binding site and a network of secondary interactions established between the inhibitor and several protein residues that are distinct from those directly involved in the anchoring of the ligand. This information can now be used to advantage in the design of more potent inhibitors.
PMID: 15317459
[Supporting
Information]
Structural Basis for the Binding of Didemnins to Human Elongation Factor eEF1A and Rationale for the Potent Antitumor Activity of These Marine Natural Products.
Marco E, Martín-Santamaría S, Cuevas C, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, and Pharma Mar S.A., Avda. de los Reyes, 1; Polígono Industrial La Mina, E-28770 Colmenar Viejo, Madrid,
Spain.
Didemnins and tamandarins are closely related marine natural products with potent inhibitory effects on protein synthesis and cell viability. On the basis of available biochemical and structural evidence and results from molecular dynamics simulations, a model is proposed that accounts for the strong and selective binding of these compounds to human elongation factor eEF1A in the presence of GTP. We suggest that the p-methoxyphenyl ring of these cyclic depsipeptides is inserted into the same pocket in eEF1A that normally lodges either the 3' terminal adenine of aminoacylated tRNA, as inferred from two prokaryotic EF-Tu·GTP·tRNA complexes, or the aromatic side chain of Phe/Tyr-163 from the nucleotide exchange factor eEF1Bα, as observed in several X-ray crystal structures of a yeast eEF1A:eEF1B complex. This pocket, which has a strong hydrophobic character, is formed by two protruding loops on the surface of eEF1A domain 2. Further stabilization of the bound depsipeptide is brought about by additional crucial interactions involving eEF1A domain 1 in such a way that the molecule fits snugly at the interface between these two domains. In the GDP-bound form of eEF1A, this binding site exists only as two separate halves, which accounts for the much greater affinity of didemnins for the GTP-bound form of this elongation factor. This binding mode is entirely different from those seen in the complexes of the homologous prokaryotic EF-Tu with kirromycin-type antibiotics or the cyclic thiazolyl peptide antibiotic GE2270A. Interestingly, the set of interactions used by didemnins to bind to eEF1A is also distinct from that used by eEF1Bα or eEF1Bβ, thus establishing a competition for binding to a common site that goes beyond simple molecular mimicry. The model presented here is consistent with both available biochemical evidence and known structure-activity relationships for these two classes of natural compounds and synthetic analogues and provides fertile ground for future research.
PMID: 15317456
| |
|
1SYW Computer model of a complex between human elongation factor eEF1A and protein synthesis inhibitor didemnin B |
Identification of the minimal conserved structure of HIV-1 protease in the presence and absence of drug pressure.
Ceccherini-Silberstein F, Erba F, Gago F, Bertoli A, Forbici F, Bellocchi MC, Gori C, D'Arrigo R, Marcon L, Balotta C, Antinori A, Monforte AD, Perno CF.
Department of Experimental Medicine, University of Rome 'Tor Vergata', Italy, the Department of Pharmacology, School of Medicine, University of Alcala, Spain, National Institute for Infectious Diseases, 'L. Spallanzani', Rome, the Department of Histology, Microbiology and Medical Biotechnologies, University of Padua, and the Institute of Infectious and Tropical Diseases, University of Milan, Italy.
OBJECTIVE: To define the extent of amino acid protease (PR) conservation in vivo in the absence and presence of pharmacological pressure in a large patient cohort. METHODS: Plasma-derived complete protein PR sequences from a well-defined cohort of 1096 HIV-1 infected individuals (457 drug-naive and 639 under antiretroviral therapy including PR-inhibitors) were obtained and analysed, and are discussed in a structural context. RESULTS: In naive patients, the PR sequence showed conservation (< 1% variability) in 68 out of 99 (69%) residues. Five large conserved regions were observed, one (P1-P9) at the N-terminal site, another (E21-V32) comprised the catalytic active-site, a third (P44-V56) contained the flap, a fourth contained the region G78-N88, and another (G94-F99) contained the C-terminal site. In PR-inhibitor treated patients, the appearance of mutations primarily associated with drug resistance determined a decrease of amino acid invariance to 45 out of 99 residues (45% conservation). The overall degree of enzyme conservation, when compared to the PR sequences in drug-naive patients, was preserved at the N- and C-terminal regions, whereas the other large conserved areas decreased to smaller domains containing, respectively, the active-site residues D25-D29, the tip of the flap G49-G52, and the G78-P81 and G86-R87 turns. CONCLUSIONS: Amino acid conservation in HIV PR can be minimally present in 45 residues out of 99. Identification of these invariable residues, with crucial roles in dimer stability, protein flexibility and catalytic activity, and their mapping on the three-dimensional structure of the enzyme will help guide the design of novel resistance-evading drugs.
PMID: 15280771
HIV protease inhibition: limited recent progress and advances in understanding current pitfalls.
Rodríguez-Barrios F, Gago F.
Department of Pharmacology, University of Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
The identification of HIV-1 protease (HIVp) as a target for therapeutic intervention against AIDS was soon followed by major efforts to understand
its substrate specificity, reaction kinetics and three-dimensional structure, both in the free state and in complex with a number of ligands including substrate mimics, products, and inhibitors. On the whole these studies have been extremely successful and have had a major impact on our understanding of ligand-receptor interactions and enzyme inhibition mechanisms. HIVp has also become a paradigm for the development and testing of new drug-design methodologies both in vitro and in silico. Even though thousands of potential HIVp inhibitors exhibiting amazing chemical diversity have been synthesized or identified from natural sources, only a few have turned out to be useful for human therapy. Although the alternative goal of preventing enzyme dimerization has been achieved as a proof of concept, this approach has not yet yielded a clinical candidate. The review covers the general strategies that led to some of the most useful inhibitors, the reasons for our limited success in effectively inhibiting this retroviral target in a clinical setting, current progress with second-generation inhibitors, and new avenues for research.
PMID: 15134553
TSAO compounds: the comprehensive story of a unique family of HIV-1 specific inhibitors of reverse transcriptase.
Camarasa MJ, San-Félix A, Velázquez S, Pérez-Pérez MJ, Gago F, Balzarini J.
Instituto de Química Médica (C.S.I.C.), Juan de la Cierva 3, 28006 Madrid, Spain. mj.camarasa@iqm.csic.es.
Emergence of drug-resistant viral strains is one of the major milestones and the main cause for the failure of antiretroviral therapy. Combination
of different anti-HIV agents has become the standard clinical practice to keep the viral load at low or even undetectable levels and to prevent
emergence of virus-drug resistance. Among the human immunodeficiency virus (HIV) reverse transcriptase (RT) inhibitors, the so called nonnucleoside RT inhibitors (NNRTIs) have gained a definitive place in the treatment of HIV infections in combination with nucleoside analogue RT inhibitors (NRTIs) and HIV protease inhibitors (PIs). The virus can be markedly suppressed for a relatively long period of time when exposed to multiple drug combination therapy (highly active antiretroviral therapy, HAART). TSAO derivatives are a peculiar group of highly functionalized nucleosides that belong to the so-called nonnucleoside RT inhibitors (NNRTIs). They exert their unique selectivity for HIV-1 through a specific interaction with the p51 subunit of HIV-1 RT. They are the first small molecules that seem to interfere with the dimerization process of the enzyme. This review covers the work carried out with this unique class of specific inhibitors of HIV-1 reverse transcriptase, including structure activity relationship studies (SAR), its mechanism of action, resistance studies, model of interaction with the enzyme, etc.
PMID: 15134550
Chemometrical Identification of Mutations in HIV-1 Reverse Transcriptase Conferring Resistance or Enhanced Sensitivity to Arylsulfonylbenzonitriles.
Rodríguez-Barrios F, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871
Alcalá de Henares, Madrid, Spain.
A Comparative Binding Energy (COMBINE) analysis on a series of non-nucleosidic reverse transcriptase inhibitors yields a QSAR model with high predictive ability and correctly identifies the effect of mutations on relevant protein residues.
PMID: 14995186
[Supporting
Information]
Benzo[f]azino[2,1-a]phthalazinium Cations: Novel DNA Intercalating Chromophores with Antiproliferative Activity.
Martínez V, Burgos C, Alvarez-Builla J, Fernández G, Domingo A, García-Nieto R, Gago F, Manzanares I, Cuevas C, Vaquero JJ.
Departamento de Química Orgánica, Departamento de Bioquímica y Biología Molecular, and Departamento de Farmacología, Universidad de Alcalá, 28871-Alcalá de Henares, Madrid, Spain, and Lab. PharmaMar, Avda. de los Reyes, 1, Polígono Industrial La Mina-Norte, 28770-Colmenar Viejo, Madrid, Spain.
New azaquinolizinium-type cations have been obtained from isochromane.
The synthesis was completed over seven steps and included as the key feature an intramolecular Westphal condensation. This first example of the intramolecular process allowed the preparation of benzo[f]pyrido[2,1-a]phthalazinium and benzo[f]quino[2,1-a]phthalazinium salts, which were evaluated as DNA intercalators, DNA topoisomerase I inhibitors, and antiproliferative compounds. Both cationic systems behave as DNA intercalators and exhibit antiproliferative activity. The pentacyclic benzo[f]quino[2,1-a]phthalazinium
cations also have an inhibitory effect on the catalytic activity of DNA topoisomerase I, without trapping of cleavage complexes. Structural characterization using density functional theory indicates that the fused ring systems are slightly nonplanar, and additional molecular modeling studies suggest a preferred orientation for the intercalating chromophores within a typical CpG or TpG intercalation site.
PMID: 14971893
Role of Histidine-85 in the Catalytic Mechanism of Thymidine Phosphorylase As Assessed by Targeted Molecular Dynamics Simulations and Quantum Mechanical Calculations.
Mendieta J, Martín-Santamaría S, Priego EM, Balzarini J,
Camarasa MJ, Pérez-Pérez MJ, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, Instituto de Química Médica, CSIC, Juan de la Cierva 3, 28006 Madrid, Spain, and Rega Institute for Medical Research, K. U. Leuven, Minderbroedersstraat 10, B-3000 Leuven,
Belgium.
The structural changes taking place in the enzyme thymidine phosphorylase (TPase, also known as PD-ECGF) that are required to achieve catalytic competence upon binding thymidine and phosphate have been simulated by means of targeted molecular dynamics (tMD). The hinge regions were characterized by structural homology comparisons with pyrimidine nucleoside phosphorylase, whose X-ray structure has been solved both in a closed and in an open form. The rearrangement of residues around the substrate that was observed during the tMD trajectory suggested that His-85 could be playing an important role in the catalytic mechanism. A quantum mechanical study of the reaction in the presence of the most relevant active site residues was then performed at the semiempirical level. The results revealed that His-85 could be involved in the protonation of the pyrimidine base at the O2 position to yield the enol tautomer of the base. To establish the role of this oxygen atom in the reaction, ground states, transition states, and final products were studied using higher level ab initio methods starting from both thymidine and 2-thiothymidine as alternative substrates. Comparison of both transition states showed that replacing the oxygen at position 2 of the pyrimidine base by sulfur should accelerate the reaction rate. Consistent with this result, 2-thiothymidine was shown to be a better substrate for TPase than the natural substrate,
thymidine. For simulating the final step of the reaction, tMD simulations were used to study domain opening upon product formation considering both
the enol and keto tautomers of thymine. Product release from the enzyme was easiest in the simulation that incorporated the keto tautomer of thymine,
suggesting that the enol intermediate spontaneously tautomerizes back to the more energetically stable keto form. These results highlight a previously unreported role for His-85 in the catalytic mechanism of TPase and can have important implications for the design of novel TPase inhibitors.
PMID: 14717594
Development of a New Family of Conformationally Restricted Peptides as Potent Nucleators of β-Turns. Design, Synthesis, Structure, and Biological Evaluation of a β-Lactam Peptide Analogue of Melanostatin.
Palomo C, Aizpurúa JM, Benito A, Miranda JI, Fratila RM, Matute C, Domercq M, Gago F, Martín-Santamaría S, Linden A.
Contribution from the Departamento de Química Orgánica I.
Facultad de Química. Universidad del País Vasco. Apdo. 1072,
20080 San Sebastián, Spain.
Novel enantiopure (i)-(β-lactam)-(Gly)-(i+3) peptide models, defined by the presence of a central α-alkyl-α-amino-β-lactam ring placed as the (i+1) residue, have been synthesized in a totally stereocontrolled way by α-alkylation of suitable N-[bis(trimethylsilyl)methyl]-β-lactams. The structural properties of these β-lactam pseudopeptides have been studied by X-ray crystallography, Molecular Dynamics simulation, and NOESY-restrained NMR simulated annealing techniques, showing a strong tendency to form stable type II or type II' β-turns either in the solid state or in highly coordinating DMSO solutions. Tetrapeptide models containing syn- or anti-α,β-dialkyl-α-amino-β-lactam rings have also been synthesized and their conformations analyzed, revealing that α-alkyl substitution is essential for β-turn stabilization. A β-lactam analogue of melanostatin (PLG amide) has also been prepared, characterized as a type-II β-turn in DMSO-d6 solution, and tested by competitive binding assay as a dopaminergic D2 modulator in rat neuron cultured cells, displaying moderate agonist activity in the micromolar concentration range. On the basis of these results, a novel peptidomimetic design concept, based on the separation of constraint and recognition elements, is proposed.
PMID: 14692766
Comparative binding energy analysis of haloalkane dehalogenase substrates: modelling of enzyme-substrate complexes by molecular docking and quantum mechanical calculations.
Kmunicek J, Bohac M, Luengo S, Gago F, Wade RC, Damborsky J.
National Centre for Biomolecular Research, Masaryk University, Kotlarska 2, 61137 Brno, Czech Republic.
We evaluate the applicability of automated molecular docking techniques and quantum mechanical calculations to the construction of a set of structures of enzyme-substrate complexes for use in Comparative binding energy (COMBINE) analysis to obtain 3D structure-activity relationships. The data set studied consists of the complexes of eighteen substrates docked within the active site of haloalkane dehalogenase (DhlA) from Xanthobacter autotrophicus GJ10. The results of the COMBINE analysis are compared with previously reported data obtained for the same dataset from modelled complexes that were based on an experimentally determined structure of the DhlA-dichloroethane complex. The quality of fit and the internal predictive power of the two COMBINE models are comparable, but better external predictions are obtained with the new approach. Both models show a similar composition of the principal components. Small differences in the relative contributions that are assigned to important residues for explaining binding affinity differences can be directly linked to structural differences in the modelled enzyme-substrate complexes: (i) rotation of all substrates in the active site about their longitudinal axis, (ii) repositioning of the ring of epihalohydrines and the halogen substituents of 1,2-dihalopropanes, and (iii) altered conformation of the long-chain molecules (halobutanes and halohexanes). For external validation, both a novel substrate not included in the training series and two different mutant proteins were used. The results obtained can be useful in the future to guide the rational engineering of substrate specificity in DhlA and other related enzymes.
PMID: 14635723
Synthesis, Activity, and Molecular Modeling Studies of Novel Human Aldose Reductase Inhibitors Based on a Marine Natural Product.
De La Fuente JA, Manzanaro S, Martin MJ, De Quesada TG, Reymundo I, Luengo SM, Gago F.
Instituto Biomar, S.A., Polígono Industrial, Edificio CEEI, 24231 Onzonilla, León, Spain; Pharma Mar, S.A., Pol. Ind. La Mina-Norte, Avda. de los Reyes 1, 28770 Colmenar Viejo, Madrid, Spain; and Departamento de Farmacología,
Universidad de Alcalá, 28871 Alcalá de Henares, Madrid, Spain.
Aldose reductase (ALR2) has been implicated in the etiology of diabetic complications, including blindness. Because of the limited number of currently available drugs for the prevention of these long-term complications, the discovery of new ALR2 inhibitors appears highly desirable. In this study, a polybrominated diphenyl ether (1) naturally occurring in a marine sponge was found to inhibit recombinant human ALR2 with an IC50 of 6.4 µM. A series of polyhalogenated analogues that were synthesized and tested in vitro to explore the structure-activity relationships displayed various degrees of inhibitory activity. The most active compounds were also capable of preventing sorbitol accumulation inside human retinal cells. In this cell-based assay, the most potent synthesized analogue (16) showed a 17-fold increase in inhibitory activity compared to that of sorbinil (IC50 = 0.24 vs 4 µM). A molecular representation of human ALR2 in complex with the natural product was built using homology modeling, automated docking, and energy refinement methods. AMBER parameters for the halogen atoms were derived and calibrated using condensed phase molecular dynamics simulations of fluorobenzene, chlorobenzene, and bromobenzene. Inhibitor binding is proposed to cause a conformational change similar to that recently reported for zenarestat. A free energy perturbation thermodynamic cycle allowed us to assess the importance of a crucial bromine atom that distinguishes the active lead compound from a much less active close natural analogue. Remarkably, the spatial location of this bromine atom is equivalent to that occupied by the only bromine atom present in zenarestat.
PMID: 14613323
[Supporting Information PDF (129.36 kB)]
Solution structure and stability of a disulfide cross-linked nucleopeptide duplex.
Gómez-Pinto I, Marchán V, Gago F, Grandas A, González C.
Instituto de Química Física Rocasolano, c/ Serrano 119, 28006
Madrid, Spain.
NMR methods are used to study the structure and stability of the duplex formed by the nucleopeptide [Ac-Cys-Gly-Ala-Hse(p3'dGCATGC)-Ala-OH]2[S-S],
in which the oligonucleotide is self-complementary and the cysteine residues of the two peptide chains form a disulfide bridge; thermal transitions
and NMR-derived structural calculations are consistent with a 3-D structure in which the oligonucleotide forms a standard B-DNA helix without significant distortions; the peptide chains are relatively disordered in solution and lie in the minor groove of the DNA helix; this nucleopeptide duplex exhibits a high melting temperature, indicating that peptide-oligonucleotide conjugates containing cysteines are suitable molecules to establish cross-links between DNA strands and stabilize the duplex.
PMID: 14594279
| |
|
1J9N Solution structure of the nucleopeptide [Ac-Lys-Trp-Lys-Hse(P3dGCATCG)-Ala]-[P5dCGTAGC] |
|
|
Nucleosides Nucleotides Nucleic Acids. 2003 May-Aug;22(5-8):951-953 |
Towards new thymidine phosphorylase/PD-ECGF inhibitors based on the transition state of the enzyme reaction.
Priego EM, Mendieta J, Gago F, Balzarini J, De Clercq E, Camarasa MJ,Pérez-Pérez MJ.
Instituto de Química Médica (C.S.I.C.), Madrid, Spain. empriego@iqm.csic.es
Computational studies have been conducted to build a closed form of TPase and to characterize the transition state of the phosphorylisis reaction
catalyzed by TPase. The results obtained point to a crucial role of His-85 and the O2 of thymine in the catalysis. This modelled transition state
forms the basis for the design of new TPase inhibitors.
PMID: 14565319
Synthesis and Structural Characterization of Pyrimidine Bi- and Tricyclic Nucleosides with Sugar Puckers Conformationally Locked into the Eastern Region of the Pseudorotational Cycle.
Cristina Chamorro, Santos M. Luengo, María-Cruz Bonache, Sonsoles Velázquez, María-Jesús Pérez-Pérez, María-José Camarasa, Federico Gago, María-Luisa Jimeno, and Ana San-Félix
Instituto de Química Médica (CSIC), Juan de la Cierva 3,
E-28006, Madrid, Spain
Reaction of 5'-O-Tosyl TSAO-m3T (1) with amines has led to the synthesis of new classes of bi- and tricyclic nucleosides. Full details about the synthesis of these compounds and a plausible mechanism to explain their obtention are reported. Additionally, we also describe the development of a second, more efficient and higher yielding synthetic route as a general approach for the synthesis of some of these bicyclic nucleosides. In order to study the conformational behaviour of the bi- and tricyclic nucleosides described in this paper, intensive NMR investigations and molecular modeling studies were performed. Conformational analysis indicates that the furanose ring of the compounds described here prefers the eastern side of the pseudorotation cycle with the base substituents preferentially in the anti-range. The torsion angle γ describing the C-4'-C-5' bond is found to prefer the +sc range. These compounds represent a novel class of E-type conformationally restricted bicyclic ribo-nucleosides containing a [3.3.0] fused carbohydrate moiety. The bicyclic nucleosides described herein present an interesting potential for diverse and selective chemical treatments on the 2'-hydroxyl and/or the functionalities incorporated at the bridge between C-3' and C-5'.
PMID: 12919035
Improving the selectivity of acyclic nucleoside analogues as inhibitors of human mitochondrial thymidine kinase: replacement of a triphenylmethoxy moiety with substituted amines and carboxamides.
Ana-Isabel Hernández, Jan Balzarini, Fátima Rodríguez-Barrios, Ana San-Félix, Anna Karlsson, Federico Gago, María-José Camarasa and María Jesús Pérez-Pérez
Instituto de Química Médica (CSIC), Juan de la Cierva 3,
E-28006, Madrid, Spain
Two series of analogues of the novel human mitochondrial thymidine kinase inhibitor 1-[(Z)-4-(triphenylmethoxy)-2-butenyl]thymine were synthesized by replacing the triphenylmethoxy moiety by a variety of substituted amines and carboxamides. In all the cases, the selectivity against the mitochondrial enzyme was either maintained or improved, and several derivatives were almost as potent as the parent compound. A molecular model was built that can account for the observed selectivities.
PMID: 12941326
Guanidinium Receptors as Enantioselective Amino Acid Membrane Carriers.
Breccia P, Van Gool M, Perez-Fernandez R, Martin-Santamaria S, Gago F, Prados P, de Mendoza J.
Contribution from the Departamento de Quimica Orgánica, Universidad Autónoma de Madrid, Cantoblanco, E-28049 Madrid, Spain, and Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
A number of artificial carriers for the transport of zwitterionic aromatic amino acids across bulk model membranes (U-tube type) have been prepared and evaluated. 1,2-Dichloroethane and dichloromethane were employed in the organic phase. All compounds are based on a bicyclic chiral guanidinium scaffold that ideally complements the carboxylate function. The guanidinium central moiety was attached to crown ethers or lasalocid A as specific subunits for ammonium recognition as well as to aromatic or hydrophobic residues to evaluate their potential interaction with the side chains of the guest amino acids. The subunits were linked to the guanidinium through ester or amide connectors. Amides were found to be better carriers than esters, though less enantioselective. On the other hand, crown ethers were superior to lasalocid derivatives. As expected, transport rates were dependent on the carrier concentration in the liquid membrane. Reciprocally, enantioselectivities were much higher at lower carrier concentrations. The results show that our previously proposed three-point binding model (J. Am. Chem. Soc. 1992, 114, 1511-1512), involving the participation of the aromatic or hydrophobic residue to interact with the side chains of the amino acid guest, is unnecessary to explain the high enantioselectivities observed. Molecular dynamics fully support a two-point model involving only the guanidinium and crown ether moieties. These molecules constitute the first examples of chiral selectors for underivatized amino acids acting as carriers under neutral conditions.
PMID: 12837099
Assessment by Molecular Dynamics Simulations of the Structural Determinants of DNA Binding Specificity for Transcription Factor Sp1.
Marco E, García-Nieto R, Gago F
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
The DNA-binding domain (DBD) of the ubiquituous transcription factor Sp1 consists of three consecutive zinc fingers that recognize a number of nucleotide sequences different from, but related to and sometimes overlapping, those recognized by the structurally better characterized early growth response protein 1 (EGR1, also known as Zif268, Krox-24, and NGFI-A). The accepted consensus binding sequence for Sp1 is usually defined by the asymmetric hexanucleotide core GGGCGG but this sequence does not include, among others, the GAG (= CTC) repeat that constitutes a high-affinity site for Sp1 binding to the wt1 promoter. Since no 3D structure of the whole DBD of Sp1 is available, either alone or in complex with DNA, a homology-based model was built and its interaction with two DNA 14-mers was studied using nanosecond molecular dynamics simulations in the presence of explicit water molecules. These oligonucleotides represent Sp1 target sites that are present in the promoters of the mdr1 and wt1 genes. For comparative purposes and validation of the protocol, the complex between the DBD of EGR1 and its DNA target site within the proximal mdr1 promoter was simulated under the same conditions. Some water molecules were seen to play an important role in recognition and stabilization of the proteinDNA complexes. Our results, which are supported by the available experimental evidence, suggest that the accuracy in the prediction of putative Sp1-binding sites can be improved by interpreting a set of rules, which are a blend of both stringency and tolerance, for the juxtaposed triplet subsites to which each zinc finger binds. Our approach can be extrapolated to WT1 and other related natural or artificial zinc-finger-containing DNA-binding proteins and may aid in the assignment of particular DNA stretches as allowed or disallowed-binding sites.
PMID: 12683994
Solution structure and stability of tryptophan-containing nucleopeptide duplexes.
Gómez-Pinto I, Marchán V, Gago F, Grandas A, González C.
Instituto de Química Física "Rocasolano" CSIC, c/ Serrano
119, 28006 Madrid, Spain, Fax: (+34) 915-642-431.
Covalently linked peptide-oligonucleotide hybrids were used as models for studying tryptophan-DNA interactions. The structure and stability of several hybrids in which peptides and oligonucleotides are linked through a phosphodiester bond between the hydroxy group of a homoserine (Hse) side chain and the 3'-end of the oligonucleotide, have been studied by both NMR and CD spectroscopy and by restrained molecular dynamics methods. The three-dimensional solution structure of the complex between Ac-Lys-Trp-Lys-Hse(p3'dGCATCG)-Ala-OH
(p=phosphate, Ac=acetyl) and its complementary strand 5'dCGTAGC has been determined from a set of 276 experimental NOE distances and 33 dihedral angle constraints. The oligonucleotide structure is a well-defined duplex that belongs to the B-form family of DNA structures. The covalently linked peptide adopts a folded structure in which the tryptophan side chain stacks against the 3'-terminal guanine moiety, which forms a cap at the end of the duplex. This stacking interaction, which resembles other tryptophan-nucleobase interactions observed in some protein-DNA complexes, is not observed in the single-stranded form of Ac-Lys-Trp-Lys-Hse(p3'dGCATCG)-Ala-OH, where
the peptide chain is completely disordered. A comparison with the pure DNA duplex, d(5'GCTACG3')-(5'CGTAGC3'), indicates that the interaction between the peptide and the DNA contributes to the stability of the nucleopeptide duplex. The different contributions that stabilize this complex have been evaluated by studying other nucleopeptide compounds with related sequences.
PMID: 12512075
[Supporting
Information]
Synthesis of 3''-Substituted TSAO Derivatives with Anti-HIV-1 and Anti-HIV-2 Activity through an Efficient Palladium-Catalyzed Cross-Coupling Approach
Esther Lobatón, Fátima Rodríguez-Barrios, Federico Gago, María-Jesús Pérez-Pérez, Erik De Clercq, Jan Balzarini, María-José Camarasa, and Sonsoles Velázquez
Instituto de Química Médica (C.S.I.C.), Juan de la Cierva 3, E-28006 Madrid, Spain, Departamento de Farmacología, Universidad
de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, and Rega Institute for Medical Research, K. U. Leuven, Minderbroedersstraat
10, B-3000 Leuven, Belgium
Various synthetic studies for the introduction of several functional groups at position 3'' of the spiro moiety of TSAO derivatives have been
explored. Among them, Stille cross-coupling of 3''-iodo-TSAO derivatives with different stannanes provided an efficient and straightforward route
for the direct and selective functionalization of the 3''-position of the sultone spiro moiety via carbon-carbon bond formation. The compounds synthesized were evaluated for their inhibitory effect on HIV-1 and HIV-2 replication in cell culture. The introduction of a bromine and particularly an iodine at the 3''-position conferred the highest anti-HIV-1 activity. In contrast, the presence at this position of (un)substituted vinyl, alkynyl, phenyl, or thienyl groups markedly diminished the anti-HIV-1 activity. Surprisingly, several of the 3''-alkenyl-substituted TSAO derivatives also gained anti-HIV-2 activity at subtoxic concentrations, an observation that is very unusual for NNRTIs and never observed before for TSAO derivatives. Finally, the anti-HIV-1 activity of some of the 3''-substituted TSAO derivatives is discussed in light of our recently proposed molecular model of interaction of TSAO derivatives with the interphase between the two subunits of HIV-1 reverse transcriptase.
PMID: 12190315
Diastereoselective reactions in glycine templates containing an ent-ardeemin fragment.
Martin-Santamaria S, Corzo-Suarez R, Avendaño C, Espada M, Gago F, Garcia-Granda S, Rzepa HS.
Department of Chemistry, Imperial College of Science, Technology and Medicine,
London SW7 2AY, United Kingdom.
Self-consistent reaction field solvation models derived from SCF-MO calculations are shown to be reliable in modeling the diastereoselectivity
of the reactions of the anion and cation derived from (4S)-2,4-dimethyl-2,4-dihydro-1H-pyrazino[2,1-b]quinazoline-3,6-dione (1) at C(1) with electrophiles and nucleophiles, respectively. The found anti/syn ratio of compound 8, which is a seco-ent-ardeemin analogue obtained by alkylation of 1 with gramine methiodide, confirms this computational model. A close similarity between the calculated geometry of the piperazine ring in the anti isomers of 1,2,4-trialkyl derivatives and that deduced from their 1H NMR (solution) and X-ray data has been also established.
PMID: 11925204
A 3·(ET743)-DNA Complex That Both Resembles an RNA-DNA Hybrid and Mimicks Zinc Finger-Induced DNA Structural Distortions
Marco E, García-Nieto R, Mendieta J, Manzanares I, Cuevas C, Gago F
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain, and Pharma Mar S.A., Cantoblanco,
28760 Tres Cantos, Madrid, Spain.
The antitumor ecteinascidin ET743 has been shown to inhibit the transcriptional activation of a number of genes at nanomolar concentrations. Cell sensitivity to subnanomolar concentrations of the drug has also been shown to specifically depend on the transcription-coupled nucleotide excision repair system. ET743 is known to bind covalently to the minor groove of a DNA double helix in regions comprising selected sets of three consecutive base pairs. Following alkylation of a central guanine, the minor groove is widened and the DNA is bent toward the major groove. We have previously shown that in the resulting adduct the DNA triplet containing the covalently modified guanine bears a strong resemblance to a DNA triplet recognized by a C2H2 zinc finger. We now expand this earlier finding and use simulation methods to show that head-to-tail binding of three ET743 molecules to three adjacent optimal binding sites stabilizes a DNA structure whose conformation is intermediate between A- and B-form DNA. Furthermore, despite the increase in roll at the sites of covalent attachment, no net curvature is apparent in this complex due to cancellation of the localized bends over virtually one turn of the helix. Both observations are in good analogy to findings in zinc finger-DNA complexes. Triplets are virtually superimposable both directly and upon shifting the register one base pair. In this latter case, the central guanine in a triplet alkylated by ET743 corresponds to the third nucleic base in the triplet recognized by a zinc finger of transcription factors such as EGR1 or Sp-1. The DNA conformation found in the ET743-DNA complex is also strongly reminiscent of an RNA-DNA hybrid, as found in
the RNA polymerase II elongation complex. The possible biological implications of these findings in relation to the antitumor action of ET743 are discussed.
PMID: 11831898
[Supporting Information]
| |
|
1KML Computer model of a covalent complex between d(GTGGCGGCGGCC) and three molecules of antitumor drug ecteinascidin 743 |
 |
Comparative Binding Energy (COMBINE) Analysis of Human Neutrophil Elastase Inhibition by Pyridone-containing Trifluoromethylketones
Cuevas C, Pastor M, Pérez C, Gago F
Departamento de Farmacología, Universidad de Alcalá, E-28871
Alcalá de Henares, Madrid, Spain
The complexes of human neutrophil elastase with a series of 40 N3-substituted trifluoromethylketone-based pyridone inhibitors have been modelled. The series spans three orders of magnitude in inhibition constants despite the fact that it was originally developed in an attempt to improve the oral activity of a lead compound. Ligand-receptor interaction energies calculated using molecular mechanics did not correlate well with the experimental activities. A good correlation with activity was found, however, when a COMBINE analysis of the same data was carried out, which allowed a quantitative interpretation of the modelled complexes. The essence of this method is to partition the ligand-receptor interaction energies into individual residue-based van der Waals and electrostatic contributions, and to subject the resulting energy matrix to partial least squares analysis. Incorporation of two additional descriptors representing the electrostatic energy contributions to the partial desolvation of both the receptor and the ligands improved the QSAR model, as did the replacement of the distance-dependent electrostatic contributions with solvent-screened electrostatic interactions calculated by numerically solving the Poisson-Boltzmann equation. The model was validated both internally
(cross-validation) and externally, using a set of twelve 6-phenyl-pyridopyrimidine analogs. The analysis reveals the subtle interplay of binding forces which occurs within the enzyme active site and provides objective information that can be interpreted in the light of the receptor structure. This information, gained from a series of real compounds, can be easily translated into 3D real or virtual database queries in the search for more active derivatives.
PMID: 11812259 |
Advances in the chemistry and pharmacology of ecteinascidins, a promising new class of anticancer agents.
Manzanares I, Cuevas C, García-Nieto R, Marco E, Gago F
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
Ecteinascidins are marine natural products consisting of two or three linked tetrahydroisoquinoline subunits and an active carbinolamine functional group. Their potent antiproliferative activity against a variety of tumor cells has made them attractive candidates for development as anticancer agents. The lead compound, ecteinascidin 743 (ET 743), is currently in phase II clinical trials but the low amounts present in its natural source, the tunicate Ecteinascidia turbinata, made it necessary to develop efficient synthetic procedures. Recent improvements on the original synthesis are reviewed as well as new strategies starting from readily available cyanosafracin B. ET 743 is known to bind to the minor groove of DNA giving rise to a covalent adduct with the exocyclic amino group at position 2 of a guanine in a fashion similar to saframycin antibiotics. Some of the resulting complexes have been studied by a variety of biochemical and spectroscopic methods and also by computer simulations. The rules for sequence specificity have been well established (preferred targets are RGC and YGG, where R and Y stand for purine and pyrimidine, respectively), and it has been shown that binding of ET 743 to DNA is accompanied by minor groove widening and DNA bending towards the major groove. Although the precise target for antitumor action remains to be unambiguously defined, a role in affecting the transcriptional
regulation of some inducible genes is rapidly emerging.
PMID: 12678757
Molecular dynamics simulations of the conformational changes of the glutamate receptor ligand-binding core in the presence of glutamate and kainate.
Mendieta J, Ramírez G, Gago F.
Centro de Biología Molecular (CSIC-UAM), Universidad Autónoma, Canto Blanco, Madrid, Spain.
Excitatory synaptic transmission is mediated by ionotropic glutamate receptors (iGluRs) through the induced transient opening of transmembrane
ion channels. The three-dimensional structure of the extracellular ligand-binding core of iGluRs shares the overall features of bacterial periplasmic binding proteins (PBPs). In both families of proteins, the ligand-binding site is arranged in two domains separated by a cleft and connected by two peptide stretches. PBPs undergo a typical hinge motion of the two domains associated with ligand binding that leads to a conformational change from an open to a closed form. The common architecture suggests a similar closing mechanism in the ligand-binding core of iGluRs induced by the binding of specific agonists. Starting from the experimentally determined kainate-bound closed form of the S1S2 GluR2 construct, we have studied by means of molecular dynamics simulations the opening motion of the ligand-binding core in the presence and in the absence of both glutamate and kainate. Our results suggest that the opening/closing interdomain hinge motions are coupled to conformational changes in the insertion region of the transmembrane segments. These changes are triggered by the interaction of the agonists with the essential Glu 209 residue. A plausible mechanism for the coupling of agonist binding to channel gating is discussed.
© 2001 Wiley-Liss, Inc.
PMID: 11484223
[Supplementary
Material]
Comparative Binding Energy Analysis of the Substrate Specificity of Haloalkane Dehalogenase from Xanthobacter autotrophicus GJ10.
Kmunicek J, Luengo S, Gago F, Ortiz AR, Wade RC, Damborsky J.
National Centre for Biomolecular Research, Masaryk University, Kotlarska 2, 611 37 Brno, Czech Republic, Department of Pharmacology, University
of Alcalá, 28871 Alcalá de Henares, Madrid, Spain, Department of Physiology and Biophysics, Mount Sinai School of Medicine, One Gustave
Levy Place, Box 1218, New York, New York 10029, and European Molecular Biology Laboratory, Meyerhofstrasse 1, 69117 Heidelberg, Germany.
Comparative binding energy (COMBINE) analysis was conducted for 18 substrates of the haloalkane dehalogenase from Xanthobacter autotrophicus GJ10 (DhlA): 1-chlorobutane, 1-chlorohexane, dichloromethane, 1,2-dichloroethane, 1,2-dichloropropane, 2-chloroethanol, epichlorohydrine, 2-chloroacetonitrile, 2-chloroacetamide, and their brominated analogues. The purpose of the COMBINE analysis was to identify the amino acid residues determining the substrate specificity of the haloalkane dehalogenase. This knowledge is essential for the tailoring of this enzyme for biotechnological applications. Complexes of the enzyme with these substrates were modeled and then refined by molecular mechanics energy minimization. The intermolecular enzyme-substrate energy was decomposed into residue-wise van der Waals and electrostatic contributions and complemented by surface area dependent and electrostatic desolvation terms. Partial least-squares projection to latent structures analysis was then used to establish relationships between the energy contributions and the experimental apparent dissociation constants. A model containing van der Waals and electrostatic intermolecular interaction energy contributions calculated using the AMBER force field explained 91% (73% cross-validated)
of the quantitative variance in the apparent dissociation constants. A model based on van der Waals intermolecular contributions from AMBER and electrostatic interactions derived from the Poisson-Boltzmann equation explained 93% (74% cross-validated) of the quantitative variance. COMBINE models predicted correctly the change in apparent dissociation constants upon single-point mutation of DhlA for six enzyme-substrate complexes.
The amino acid residues contributing most significantly to the substrate specificity of DhlA were identified; they include Asp124, Trp125, Phe164,
Phe172, Trp175, Phe222, Pro223, and Leu263. These residues are suitable targets for modification by site-directed mutagenesis.
PMID: 11467952
[Corrigenda][Supporting
Information]
Exploring the role of the 5'-position of TSAO-T. Synthesis and anti-HIV evaluation of novel TSAO-T derivatives.
Chamorro C, Pérez-Pérez M, Rodriguez-Barrios F, Gago F, De Clercq E, Balzarini J, San-Felix A, Camarasa M.
Instituto de Química Médica (C.S.I.C.), c/ Juan de la Cierva 3, E-28006, Madrid, Spain
Various analogues of the anti-HIV-1 agent TSAO-T, [1-[2',5'-bis-O-(tert-butyldimethylsilyl)-&beta-D-ribofuranosyl]thymine]-3'-spiro-5"-(4"-amino-1",2"-oxathiole-2",2"-dioxide) have been synthesized in which the 5'-TBDMS group has been replaced by alkyl-, alkenyl- or aromatic ether groups, substituted amines, carbamoyl or (thio)acyl groups. The compounds synthesized were evaluated for their inhibitory effect on HIV-1 and HIV-2 replication in cell culture. Replacement of the 5'-TBDMS group by an acyl, aromatic or a cyclic moiety markedly diminish or even eliminate the anti-HIV activity. However, the presence at that position of an alkyl or alkenyl chain, partially retain antiviral activity. These observations suggest that the 5'-TBDMS group of the TSAO molecule plays a crucial role.
PMID: 11397508
Identification of a putative binding site for [2',5'-bis-O-(tert-butyldimethyl-silyl)-β-D-ribofuranosyl]-3'-spiro-5''-(4''-amino-1'',2''-oxathiole-2'',2''-dioxide)thymine (TSAO) derivatives at the p51-p66 interface of HIV-1 reverse transcriptase.
Rodríguez-Barrios F, Pérez C, Lobatón E, Velázquez S, Chamorro C, San-Félix A, Pérez-Pérez MJ, Camarasa MJ, Pelemans H, Balzarini J, Gago F.
Departamento de Farmacología, Universidad de Alcalá, E-28871 Alcalá de Henares, Madrid, Spain.
A binding site for TSAO-m3T at the interface between the p66 and p51 subunits of HIV-1 reverse transcriptase (RT) and distinct from that of
"classical" HIV-1 non-nucleoside inhibitors is proposed. The feasibility of the binding mode was assessed by carrying out nanosecond molecular dynamics simulations for the complexes of TSAO-m3T with reduced models of both the wild-type enzyme and a more sensitive R172A mutant. The molecular model is in agreement with a previous proposal, with known structure-activity and mutagenesis data for this unique class of inhibitors, and also with recent biochemical evidence indicating that TSAO analogues can affect enzyme dimerization. The relative importance of residues involved in dimer formation and TSAO-RT complex stabilization was assessed by a combination of surface area accessibility, molecular mechanics, and continuum electrostatics calculations. A structure-based modification introduced into the lead compound yielded a new derivative with improved antiviral activity.
PMID: 11384232
[Supporting Information]
Increased DNA Binding Specificity for Antitumor Ecteinascidin 743 through Protein-DNA Interactions?
García-Nieto R, Manzanares I, Cuevas C, Gago F
Departamento de Farmacología, Universidad de Alcalá, E-28871 Madrid, Spain, and Pharma Mar, Tres Cantos, E-28760 Madrid, Spain.
Ecteinascidin 743 (ET743) is a potent anticancer agent of natural origin currently undergoing phase II clinical trials. ET743 binds to the DNA minor groove following well-defined hydrogen-bonding rules and alkylates the exocyclic amino group of the central guanine in triplets such as AGC, CGG or TGG. As a consequence, widening of the minor groove and bending towards the major groove have been reported but the precise mechanism by which
ET743 exerts its anticancer activity has not been elucidated yet. We now show that the minor grooves of DNA when bound to the zinc fingers of transcription factor EGR-1/Zif268 and in the covalent complex with ET743 are virtually superimposable. This finding suggests that the unusually high potency and unique antiproliferative mechanism of ecteinascidin-like molecules may depend on their binding to a DNA stretch that becomes pre-organized and structurally complementary to the wedge shape of the drug molecule upon binding of a specific protein. Likely candidates are members of the family of mammalian Sp/XKLF transcription factors represented by Sp1. The present hypothesis can be tested and opens new avenues for research.
PMID: 11087561
Related article:
García-Nieto, R., Manzanares,I, Cuevas,C., Gago, F. "Bending of DNA upon Binding of Ecteinascidin 743 and Phthalascidin 650 Studied by Unrestrained Molecular Dynamics Simulations"
Journal of the American Chemical Society, 122(30): 7172-7182 (2000)
| |
|
1EZ5 Computer model of a covalent complex between d(TAAAGCTTA) and antitumor drug ecteinascidin 743 |
|
1EZH Computer model of a covalent complex between d(TAACGGTTA) and antitumor drug ecteinascidin 743 |
3D-QSAR methods on the basis of ligand-receptor complexes. Application of COMBINE and GRID/GOLPE methodologies to a series of CYP1A2 ligands.
Lozano JJ, Pastor M, Cruciani G, Gaedt K, Centeno NB, Gago F, Sanz F
Research Group on Medical Informatics, IMIM, Barcelona, Spain.
Many heterocyclic amines (HCA) present in cooked food exert a genotoxic activity when they are metabolised (N-oxidated) by the human cytochrome
P450 1A2 (CYP1A2h). In order to rationalize the observed differences in activity of this enzyme on a series of 12 HCA, 3D-QSAR methods were applied
on the basis of models of HCA-CYP1A2h complexes. The CYP1A2h enzyme model has been previously reported and was built by homology modeling based on
cytochrome P450 BM3. The complexes were automatically generated applying the AUTODOCK software and refined using AMBER. A COMBINE analysis on the
complexes identified the most important enzyme-ligand interactions that account for the differences in activity within the series. A GRID/GOLPE
analysis was then performed on just the ligands, in the conformations and orientations found in the modeled complexes. The results from both methods
were concordant and confirmed the advantages of incorporating structural information from series of ligand-receptor complexes into 3D-QSAR methodologies.
PMID: 10815771, UI: 20273193
Automated docking and molecular dynamics simulations of nimesulide in the cyclooxygenase active site of human prostaglandin-endoperoxide synthase-2 (COX-2).
García-Nieto R, Pérez C, Gago F
Departamento de Farmacología, Universidad de Alcalá, Madrid,
Spain.
Molecular models of the complex between the selective COX-2 inhibitor nimesulide and the cyclooxygenase active site of human prostaglandin-endoperoxide synthase-2 have been built using a combination of homology modelling, conformational searching and automated docking techniques. The stability of the resulting complexes has been assessed by molecular dynamics simulations and interaction energy decomposition. It is found that nimesulide exploits the extra space made available by the replacement at position 523 of an isoleucine residue in COX-1 by a valine in COX-2 and establishes electrostatic interactions with both Arg-106 and Arg-499 (Arg-120 and Arg-513 in PGHS-1 numbering). Two alternate binding modes are proposed which are compatible with the pharmacological profile of this agent as a COX-2 selective inhibitor.
PMID:
10721503, UI: 20186281
Recognition elements that determine affinity and sequence-specific binding to DNA of 2QN, a biosynthetic bis-quinoline analogue of echinomycin.
Bailly C, Echepare S, Gago F, Waring MJ
Laboratoire de Pharmacologie Antitumorale, Centre Oscar Lambret, Lille, France. bailly@lille.inserm.fr
Footprinting experiments with DNase I provide a starting-point for investigating the molecular basis of nucleotide sequence recognition by 2QN, a bis-quinoline derivative of the quinoxaline antibiotic echinomycin produced by directed biosynthesis in Streptomyces echinatus. Using tyrT DNA molecules variously substituted with inosine and/or 2,6-diaminopurine residues it is shown that the location of the 2-amino group of purine nucleotides in the minor groove of the double helix exerts a dominant influence in determining where the antibiotic will bind, as it does for echinomycin. However, newly created binding sites in DNA molecules substituted with diaminopurine (D), all located round TpD steps, bind 2QN with so much higher affinity than the canonical CpG steps that the latter fail completely to appear as footprints in D-substituted DNA; indeed CpG sequences appear in regions of enhanced susceptibility to nuclease cleavage as do CpI steps in doubly D + I-substituted DNA. Quantitative footprinting plots confirm that sequences surrounding TpD steps bind 2QN several hundred-fold more tightly than do CpG-containing sequences, with dissociation constants of the order of 25 nM. To test the hypothesis that differences in stacking interactions between the chromophores of the drug and the DNA base pairs could account for the differences in binding affinities, models of 2QN bound to two DNA hexamers containing either a central CpG or a central TpD step were built. Calculation of the molecular electrostatic potential (MEP) of 2QN in solution using a continuum method revealed a distinctive pattern that is considered relevant to DNA binding. When the MEPs calculated for the two DNA hexamers in the complexed
state were compared, substantial differences were found in the major groove and in the space between the base pairs that is occupied by the chromophores of the drug upon binding. The modelling data support the notion that electrostatic stacking interactions underlie the considerably preferred binding of echinomycin and 2QN around TpD steps rather than CpG steps.
PMID: 10500504, UI: 99430291
Regulation of cyclooxygenase activity by metamizol.
Campos C, de Gregorio R, García-Nieto R, Gago F, Ortiz P, Alemany S
Medical Department, Boehringer Ingelheim, Spain.
The ability of metamizol to inhibit cyclooxygenase-1 and cyclooxygenase-2 activities has been evaluated using different cyclooxygenase sources. Metamizol inhibited purified cyclooxygenase-1 and cyclooxygenase-2 with an IC50 of about 150 µg/ml. A similar IC50 value for cyclooxygenase-2 was obtained in lipopolysaccharide-activated broken murine macrophages. Consistent with these findings, molecular models of the complexes between cyclooxygenase-1 or cyclooxygenase-2 with 4-methylaminoantipyrine, the major active derivative of metamizol, suggested a common binding mode to both isoforms. In intact cells, however, the inhibition profiles were markedly different. The IC50 values of metamizol for cyclooxygenase-1 in intact bovine aortic endothelial cells (BAEC) cells and human platelets were 1730 +/- 150 µg/ml and 486 +/- 56 µg/ml, respectively. Inhibition of cyclooxygenase-2 activity in murine macrophages and primary human leukocytes activated by lipopolysaccharide yielded IC50 values of 12 +/- 1.8 µg/ml and 21 +/- 2.9 µg/ml, respectively. These data indicate that the IC50 values obtained with purified enzymes or disrupted cells cannot always be extrapolated to the cyclooxygenase inhibitory activity of nonsteroidal antiinflammatory drugs (NSAIDs) in intact cells. The data presented here also indicate that cyclooxygenase-2 inhibition could play an important role in the pharmacological effects of metamizol.
PMID: 10493111, UI: 99420854
Targeting HIV protease: From peptidomimetics to non-peptide inhibitors.
Gago F
Department of Pharmacology, University of Alcala, E-28871 Alcala de Henares, Madrid, Spain. fgago@fisfar.alcala.es.
The identification of HIV-1 protease as a target for therapeutic intervention against AIDS, soon followed by the resolution of its three-dimensional structure, has had a major impact on drug-design methodologies. The possible HIV-1 protease inhibitors that have been synthesized number in the thousands and exhibit amazing chemical diversity, but only a few happen to be useful for human therapy. This review covers the development of some of these inhibitors, the reasons for this limited success, current therapeutic problems and challenges remaining ahead.
PMID: 16158350
Molecular model of the interaction between nimesulide and human cyclooxygenase-2.
García-Nieto R, Pérez C, Checa A, Gago F
Departamento de Farmacología, Universidad de Alcalá, Madrid, Spain.
The cyclooxygenase-2 (COX-2) isoenzyme is a key target for COX-2-selective non-steroidal anti-inflammatory drugs (NSAIDs). An important difference
in binding of nimesulide compared with non-selective NSAIDs appears to involve the amino acid at position 523 of the enzyme. Replacement of valine
with isoleucine at this position provides access to a binding site that is larger in COX-2 than in COX-1. Nimesulide appears to exploit this enlarged
binding site for establishing a number of favourable contacts with the enzyme that lead to selective inhibition of COX-2. We made these conclusions
from a three-dimensional molecular model of the active site of human COX-2, constructed using the X-ray coordinates of COX-1 from sheep seminal vesicles and COX-2 from mouse fibroblasts as templates, with the aid of sequence alignment methods and molecular modelling techniques. The resulting model was refined, and the active site was probed for regions of steric and electrostatic complementarity for ligand binding. Docking studies were then undertaken with many different nimesulide conformers, a family of which could establish very favourable interactions with the NSAID binding site of human COX-2 by exploiting the extra space made available by the isoleucine/valine replacement. The stability of the resulting complexes was studied by simulating molecular dynamics.
PMID: 10369401, UI: 99296075
Solution structure determination by two-dimensional 1H NMR of omega-conotoxin MVIID, a calcium channel blocker peptide.
Civera C, Vázquez A, Sevilla JM, Bruix M, Gago F, García AG, Sevilla P
Dpto. Química Física II, Facultad de Farmacia, UCM, Madrid, 28040, Spain.
The three-dimensional structure of omega-conotoxin MVIID has been determined in aqueous solution by two-dimensional 1H NMR techniques. A total of 267 relevant upper-bound distance restraints were used to obtain a family of convergent structures using molecular dynamics methods. A standard simulated annealing protocol using the XPLOR program included in ARIA provided a total of 18 final structures. The averaged RMSD between these structures and the mean atomic coordinates was 0.8 +/- 0.3 Å for the backbone atoms. The highest mobility was observed in the segments between residues 10 to 13, comprising Tyr 13, one of the residues shown to be important for binding of omega-conotoxin GVIA and MVIIA to N-type calcium channels. The three-dimensional structure is stabilised by the three disulfide bonds and includes a short antiparallel β-strand between residues 5-8, 23-25 and 19-21. The folding for this non-N-type calcium channel blocker is similar to that previously calculated for omega-conotoxins GVIA, MVIIA and MVIIC. This suggests the disulfide bond pattern fixes the structure. The reported three-dimensional information can be used to advantage in order to highlight the structural parameters involved in discrimination among calcium channel subtypes.
© 1999 Academic Press.
PMID: 9920728, UI: 99121185
Abasic analogues of TSAO-T as the first sugar derivatives that specifically inhibit HIV-1 reverse transcriptase.
Velázquez S, Chamorro C, Pérez-Pérez MJ, Alvarez R, Jimeno ML, Martín-Domenech A, Pérez C, Gago F, De Clercq E, Balzarini J, San-Félix A, Camarasa MJ
Instituto de Química Médica, C.S.I.C., Juan de la Cierva 3, 28006 Madrid, Spain, Departamento de Farmacología, Universidad
de Alcalá, 28871 Alcalá de Henares, Madrid, Spain, and Rega Institute for Medical Research, Katholieke Universiteit Leuv.
With the aim of assessing the role that the thymine base of TSAO-T may play in the interaction of TSAO compounds with HIV-1 reverse transcriptase
(RT), we have designed, synthesized, and evaluated for their anti-HIV-1 activity a series of 3-spiro sugar derivatives substituted at the anomeric
position with nonaromatic rings or with amine, amide, urea, or thiourea moieties that mimic parts or the whole thymine base of TSAO-T. Also, a
dihydrouracil TSAO analogue and O-glycosyl 3-spiro sugar derivatives substituted at the anomeric position with methyloxy or benzyloxy groups have been prepared. Compounds substituted at the anomeric position with an azido, amino, or methoxy group, respectively, were devoid of marked antiviral activity (EC50: 10-200 µM). However, the substituted urea sugar derivatives led to an increase in antiviral potency (EC50: 0.35-4 µM), among them those urea derivatives that mimic most closely the intact TSAO-T molecule retained the highest antiviral activity. Also, the dihydrouracil TSAO derivative retained pronounced anti-HIV-1 activity. None of the compounds showed any anti-HIV-2 activity. The results described herein represent the first examples of sugar derivatives that interact in a specific manner with HIV-1 RT. Molecular modeling studies carried out with a prototype urea derivative indicate that a heteroaromatic ring is not an absolute requirement for a favorable interaction between TSAO-T and HIV-1 RT. Urea derivatives, which can mimic to a large extent both the shape and the electrostatic potential of a thymine ring, can effectively replace this nucleic acid base when incorporated into a TSAO molecular framework with only moderate loss of activity.
PMID: 9804703, UI: 99024045
 |
Antivir Chem Chemother 1998 Nov;9(6):481-9 |
Regiospecific synthesis and anti-human immunodeficiency virus activity of novel 5-substituted N-alkylcarbamoyl and N,N-dialkylcarbamoyl 1,2,3-triazole-TSAO analogues.
Velázquez S, Alvarez R, Perez C, Gago F, De Clercq E, Balzarini J, Camarasa MJ
Instituto de Química Médica (CSIC), Madrid, Spain.
Several 5-N-alkyl and 5-N,N-dialkylcarbamoyl substituted analogues of the anti-human immunodeficiency virus (HIV) type 1 lead compound [1-[2',5'-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-5-(N,N-dimethylcarbamoyl)-1,2,3-triazole]-3'-spiro-5"-(4"-amino-1",2"- oxathiole-2",2"-dioxide) have been prepared and evaluated as inhibitors of HIV-1 replication. A new regiospecific synthetic procedure is described. The compounds were prepared by cycloaddition of the appropriate glycosylazide to 2-oxoalkylidentriphenyl-phosphoranes, followed by treatment with primary or secondary amines, to yield, exclusively, 5-substituted 1,2,3-triazole-TSAO analogues. Several 5-substituted 1,2,3-triazole-TSAO derivatives proved to be potent inhibitors of HIV-1 replication with higher antiviral selectivity than that of the parent TSAO prototype.
PMID: 9865386, UI: 99082905
|
Antivir Chem Chemother 1998 Jul;9(4):333-40 |
Novel 3'-spiro nucleoside analogues of TSAO-T. Part II. A comparative study based on NMR conformational analysis in solution and theoretical calculations.
Alvarez R, Jimeno ML, Gago F, Balzarini J, Pérez-Pérez MJ, Camarasa MJ
Instituto de Química Médica (CSIC), Madrid, Spain.
The structures of two novel 3'-spiro nucleosides analogues of the potent human immunodeficiency virus type 1 (HIV-1) reverse trancriptase (RT) inhibitor TSAO-m3T, in solution, as derived from NMR spectroscopy are described. In these TSAO analogues the spiro amino oxathioledioxide moiety has been replaced by spiro amino oxazolone or spiro amino oxathiazoledioxide moieties. A comparative study based on theoretical calculations of the hydrophobicity, the solvation free energies and molecular electrostatic potentials (MEP) of the three compounds is also described. No significant conformational differences were detected in solution between TSAO-m3T and its analogues that might account for the differences observed in their inhibitory activity against HIV-1 RT. The calculated hydrophobicity (log P) values, dipole moments and the electrostatic contributions to the solvation free energies of the three spiro ring systems were also similar. However, the differences found in the calculated MEPs of the spiro systems between TSAO-m3T and its analogues suggest that the different electrostatic surroundings of the 4"-amino group of the spiro moiety in the analogues may be responsible for a detrimental electrostatic interaction of the spiro rings with the Glu-B138 of RT.
PMID: 9875412, UI: 99092571
Stacking interactions and intercalative DNA binding.
Gago F
Departamento de Farmacología, Universidad de Alcalá, Madrid, Spain.
The DNA-binding properties of many ligands can be rationalized on the basis of their structural and electronic complementarity with the functional
groups present in the minor and major grooves of particular DNA sequences. Specific hydrogen bonding patterns are particularly useful for the purpose
of sequence recognition. Less obvious, however, is the influence of base composition on the conformational preferences of individual base steps and on the binding of intercalating moieties which become sandwiched between contiguous base pairs. Improved knowledge of stacking interactions may
lead to a better understanding of the architecture and inherent flexibility of particular DNA sequences and may provide insight into the principles
that dictate the structural changes and specificity patterns observed in the binding of some intercalating ligands to DNA.
PMID: 9571084, UI: 98249509
Comparative Binding Energy Analysis of HIV-1 Protease Inhibitors: Incorporation of Solvent Effects and Validation as a Powerful Tool in Receptor-Based Drug Design.
Pérez C, Pastor M, Ortiz AR, Gago F
Departamento de Farmacología, Universidad de Alcalá, Madrid, Spain.
A comparative binding energy (COMBINE) analysis (Ortiz et al. J. Med. Chem. 1995, 38, 2681-2691) has been performed on a training set of 33 HIV-1 protease inhibitors, and the resulting regression models have been validated using an additional external set of 16 inhibitors. This data set was originally reported by Holloway et al. (J. Med. Chem. 1995, 38, 305-317), who showed the usefulness of molecular mechanics interaction energies for predicting the activity of novel HIV-1 protease inhibitors within the framework of the MM2X force field and linear regression techniques. We first used the AMBER force field on the same set of three-dimensional structures to check up on any possible force-field dependencies. In agreement with the previous findings, the calculated raw ligand-receptor interaction energies were highly correlated with the inhibitory activities (r2 = 0.81), and the linear regression model relating both magnitudes had an acceptable predictive ability both in internal validation tests (q2 = 0.79, SDEPcv = 0.61) and when applied to the external set of 16 different inhibitors (SDEPex = 1.08). When the interaction energies were further analyzed using the COMBINE formalism, the resulting PLS model showed improved fitting properties (r2 = 0.89) and provided better estimations for the activity of the compounds in the external data set (SDEPex = 0.83). Computation of the electrostatic part of the ligand-receptor interactions by numerically solving the Poisson-Boltzmann equation did not improve the quality of the linear regression model. On the contrary, incorporation of the solvent-screened residue-based electrostatic interactions and two additional descriptors representing the electrostatic energy contributions to the partial desolvation of both the ligands and the receptor resulted in a COMBINE model that achieved a remarkable predictive ability, as assessed by both internal (q2 = 0.73, SDEPcv = 0.69) and external validation tests (SDEPex = 0.59). Finally, when all the inhibitors studied were merged into a single expanded set, a new model was obtained that explained 91% of the variance in biological activity (r2 = 0.91), with very high predictive ability (q2 = 0.81, SDEPcv = 0.66). In addition, the COMBINE analysis provided valuable information about the relative importance of the contributions to the activity of individual residues that can be fruitfully used to design better inhibitors. All in all, COMBINE analysis is validated as a powerful methodology for predicting binding affinities and pharmacological activities of congeneric ligands that bind to a common receptor.
PMID: 9526559, UI: 98187307
[Figure 5 in color (PDF)]
Simulation of alternative binding modes in a structure-based QSAR study of HIV-1 protease inhibitors.
Pastor M, Pérez C, Gago F
Department of Pharmacology, University of Alcala, Spain.
We have used a published set of inhibitors of HIV-1 protease to build a COMBINE-type structure-based QSAR model with good predictive ability
(r2 = 0.90, q2 = 0.69). Since the compounds in the training series exhibit most of their structural variability on one-half of the pseudosymmetrical binding cavity and only one binding orientation was explored for each molecule, the model describes mainly the effect of the structural changes on interactions involving only one-half of the binding cavity (pockets S1' and S2'). Thus, the model cannot be expected to give accurate predictions for new compounds exhibiting structural variation in both halves. The model does in fact show a tendency to underpredict slightly the biological activity of the molecules in the external test set. In an attempt to improve the quality of the model, both possible orientations of the ligands are now considered so that structural variation takes place in all binding pockets. One possibility would have been to build an additional set of complexes with the inhibitors docked in a reversed orientation. The alternative we have explored, however, consists of manipulating the data matrix describing the interaction energies so that each row is duplicated and the order of the variables in the duplicated rows is swapped between subunits. This simple approach has produced a new model that is similar in quality to the original model (r2 = 0.89, q2 = 0.64) but lacks the tendency to underpredict the activity of the compounds in the external set. Moreover, since equivalent residues are assigned equivalent weights, the model is insensitive to ligand orientation and is easier to interpret.
PMID: 9704299, UI: 98369922
Assessment of solvation effects on calculated binding affinity differences: trypsin inhibition by flavonoids as a model system for congeneric series.
Checa A, Ortiz AR, de Pascual-Teresa B, Gago F
Departamento de Farmacología, Universidad de Alcalá, Alcalá de Henares, Madrid, Spain.
On the basis of molecular models of the interaction between trypsin and a series of seven structurally congeneric bioflavonoid inhibitors, the influence of solvation effects in the calculation of binding free energy differences in congeneric series has been assessed. The models were derived
by making use of the X-ray crystal structure of bovine trypsin and the DOCK program, and the complementarity of the interactions between the functional groups of the docked molecules and the binding site region was corroborated independently with the GRID program. Interaction energies calculated for the complexes using molecular mechanics were found to correlate with the experimental inhibitory activities, although the quality of the correlation was dependent on the molecular modeling protocol. To understand why such correlations could be obtained in the absence of an explicit description of solvent effects, the in vitro activities were transformed into binding free energies, and continuum electrostatic theory was used to incorporate solvent effects by approximating them to the electrostatic contribution to the binding free energies. The results of our calculations show that, within this congeneric series, the cost in electrostatic free energy of desolvating both the enzyme binding site and the buried part of the inhibitors (ΔGdesolv) is roughly constant within the series. On the other hand, the electrostatic interaction energy (EeleLR) varies only slightly along the series in comparison with the van der Waals interaction (EVDWLR), and this variation is mostly solvent-independent, i.e., the reaction field energy of the solvent in the bound state (EsrfLR) makes almost a negligible contribution to the binding free energy differences. As a result, differences in binding free energy are dominated by the van der Waals term, while the electrostatic contribution is, to a good approximation, solvent-independent. A similar scenario may account for the good correlations frequently found between ligand activities and ligand-receptor interaction energies derived using plain molecular mechanics, although generality remains to be determined.
PMID: 9406602, UI: 98069700
Reliability of comparative molecular field analysis models: effects of data scaling and variable selection using a set of human synovial fluid phospholipase A2 inhibitors.
Ortiz AR, Pastor M, Palomer A, Cruciani G, Gago F, Wade RC
European Molecular Biology Laboratory, Heidelberg, Germany / Departamento de Farmacología, Universidad de Alcalá, Alcalá de
Henares, Madrid, Spain.
The effects of data pretreatment, data scaling, and variable selection on three-dimensional quantitative structure-activity relationships derived
by comparative molecular field analysis (CoMFA) using the GRID energy funcion were studied in detail for a set of inhibitors of the human synovial fluid phospholipase A2 (HSF-PLA2). The quality of the models was evaluated for predictive power and ability to map the receptor binding site by (a) comparison of predicted and experimental activities using cross-validation and external validation sets and (b) comparison of the regions selected in space in the CoMFA models with a crystal structure of a HSF-PLA2-inhibitor complex, with optimized comparative binding energy analysis (COMBINE) models (Ortiz et al., 1995) and with structure-activity relationships derived previously for different sets of compounds. It is found that (1) data scaling and dielectric modeling strongly influence CoMFA results. Unscaled data and a uniform dielectric constant of 4 are well suited to GRID-CoMFA studies for the present compound set. (2) The GOLPE and Q2-GRS variable selection methods select variables in roughly the same regions in Cartesian space, but they produce different models in chemometric space and differ in their sensitivity to data scaling and pretreatment and their tendency to overfitting. (3) CoMFA models are consistent with COMBINE models in that they identify approximately the same intermolecular interactions as relevant for activity. Our study provides support for the qualitative receptor-mapping properties of CoMFA models and for the validity of variable selection when applied with care and also provides guidelines for how to evaluate the quality of CoMFA models.
PMID: 9089335, UI: 97244495
[Supporting
Information] [Corrigenda]
Structure-affinity relationships for the binding of actinomycin D to DNA.
Gallego J, Ortiz AR, de Pascual-Teresa B, Gago F
Department of Physiology and Pharmacology, University of Alcalá, Madrid, Spain.
Molecular models of the complexes between actinomycin D and 14 different DNA hexamers were built based on the X-ray crystal structure of the actinomycin-d(GAAGCTTC)2 complex. The DNA sequences included the canonical GpC binding step flanked by different base pairs, nonclassical binding sites such as GpG and GpT, and sites containing 2,6-diamino-purine. A good correlation was found between the intermolecular interaction energies calculated for the refined complexes and the relative preferences of actinomycin binding to standard and modified DNA. A detailed energy decomposition into van der Waals and electrostatic components for the interactions between the DNA base pairs and either the chromophore or the peptidic part of the antibiotic was performed for each complex. The resulting energy matrix was then subjected to principal component analysis, which showed that actinomycin D discriminates among different DNA sequences by an interplay of hydrogen bonding and stacking interactions. The structure-affinity relationships for this important antitumor drug are thus rationalized and may be used to advantage in design of novel sequence-specific DNA-binding agents.
PMID: 9089429, UI: 97244653
Molecular dynamics simulations of the bis-intercalated complexes of ditercalinium and Flexi-Di with the hexanucleotide d(GCGCGC)2: theoretical analysis of the interaction and rationale for the sequence binding specificity.
de Pascual-Teresa B, Gallego J, Ortiz AR, Gago F
Departamento de Fisiología y Farmacología, Universidad de Alcalá, Madrid, Spain.
The X-ray crystal structures of the complexes of ditercalinium and Flexi-Di with d(CGCG)2 have been studied by computational chemistry methods in an attempt to rationalize their distinct structural features. In addition, the complexes of these two bisintercalating drugs with d(GCGCGC)2 have been modeled and subjected to 0.5 ns of molecular dynamics simulations in explicit solvent with the aim of evaluating the relative importance of hydrogen bonding and stacking interactions in the sequence binding specificity of these compounds. According to our calculations, the electrostatic term is attractive for the stacking interactions between the pyridocarbazole chromophores of these drugs and the base pairs that make up the sandwiched GpC step. On the contrary, this energy term is repulsive for the base pairs that make up the boundaries of the bisintercalation site. This differential electrostatic binding energy component, which is shown to have a strong orientational dependence, could lie at the origin of the observed binding preferences of these drugs. In addition, both the Lennard-Jones and the electrostatic energy terms contribute to stabilizing the underwound central GpC step. The attractive electrostatic interactions between the linkers and the major groove are in concert with the stacking specificities for the sandwiched GpC step, which is thus very effectively stapled by the drugs. The hydrogen-bonding potential of the linkers, however, appears to be reduced in an aqueous medium due to competing interactions with water. Binding of either ditercalinium or Flexi-Di to d(GCGCGC)2 appears to favor the A-type conformation that this DNA molecule most likely adopts in the free state. The possible relevance of these findings to the process of
bis-intercalation and to the pharmacological action of these compounds is discussed.
PMID: 8941395, UI: 97096371
[Supporting
Information]
Prediction of Drug Binding Affinities by Comparative Binding Energy Analysis.
Ortiz AR, Pisabarro MT, Gago F, Wade RC
European Molecular Biology Laboratory, Heidelberg, Germany / Departamento de Fisiología y Farmacología, Universidad de Alcalá,
Alcalá de Henares, Madrid, Spain.
A new computational method for deducing quantitative structure-activity relationships (QSARs) using structural data from ligand-macromolecule complexes is presented. First, the ligand-macromolecule interaction energy is computed for a set of ligands using molecular mechanics calculations. Then, by selecting and scaling components of the ligand-macromolecule interaction energy that show good predictive ability, a regression equation is obtained in which activity is correlated with the interaction energies of parts of the ligands and key regions of the macromolecule. Application to the interaction of the human synovial fluid phospholipase A2 with 26 inhibitors indicates that the derived QSAR has good predictive ability and provides insight into the mechanism of enzyme inhibition. The method, which we term comparative binding energy (COMBINE) analysis, is expected to be applicable to ligand-receptor interactions in a range of contexts including rational drug design, host-guest systems, and protein engineering.
PMID: 7629807, UI: 95356160
Erratum in J Med Chem 1997 Dec 5;40(25):4168
Protein Eng 1994 Dec;7(12):1455-1462
Molecular modeling of the interaction of polyproline-based peptides with the Abl-SH3 domain: rational modification of the interaction.
Pisabarro MT, Ortiz AR, Viguera AR, Gago F, Serrano L
European Molecular Biology Laboratory, Heidelberg, Germany / Departamento de Fisiología y Farmacología, Universidad de Alcalá, Alcalá de Henares, Madrid, Spain.
A molecular model of the interaction of polyproline-rich peptides with the Abl-SH3 domain is proposed, based on docking calculations with the
DOCK program coupled with molecular dynamics simulations. Two distinct binding modes of the peptide to the same aromatic-rich region (Tyr10, Phe12,
Trp39, Trp50, Tyr55) of the domain were obtained. It is proposed that these two models could represent different binding modes of proline-rich peptides to Src homology region 3 domains. Several peptide mutants were designed to determine whether the two orientations were possible. Analysis of the Kd values and fluorescence emission of these peptides indicate that one of the orientations is more plausible and that residues at position 4 of the peptide interact with the RT loop, being important in modulating the peptide affinity for the Abl-SH3 domain.
PMID: 7716156, UI: 95232108
Binding of echinomycin to d(GCGC)2 and d(CCGG)2: distinct stacking interactions dictate the sequence-dependent formation of Hoogsteen base pairs.
Gallego J, Luque FJ, Orozco M, Gago F
Departamento de Fisiología y Farmacología, Universidad de Alcalá de Henares, Madrid, Spain.
Molecular dynamics simulations have been used to explore the behavior of the complexes of echinomycin with the DNA tetramers d(GCGC)2 and d(CCGG)2 in which the terminal bases have been paired according to either a Hoogsteen or a Watson-Crick hydrogen bonding scheme. The energy of the four resulting complexes has been monitored along the dynamics trajectories and the interaction energy between echinomycin and DNA has been decomposed into contributions arising from the planar aromatic systems and the depsipeptide part of the antibiotic. Our calculations predict a large increase in overall stabilization upon protonation of the terminal cytosines and subsequent Hoogsteen pair formation in the complex of echinomycin with d(GCGC)2 but not with d(CCGG)2, in agreement with the experimental evidence [Gao and Patel, Quart. Rev. Biophys. 22, 93-138 (1989)]. The conformational preferences appear to arise mainly from differential stacking interactions in which the electrostatic component is shown to play a dominant role. Differences in hydrogen bonding patterns are also found among the complexes and these are compared in relation to available crystal structures. The binding of echinomycin to DNA appears as a complex process involving many interrelated variables.
PMID: 7848562, UI: 95151198
DNA sequence-specific reading by echinomycin: role of hydrogen bonding and stacking interactions.
Gallego J, Luque FJ, Orozco M, Burgos C, Alvarez-Builla J, Rodrigo MM, Gago F
Departamento de Fisiología y Farmacología, Universidad de Alcalá de Henares, Madrid, Spain.
The binding of echinomycin to DNA hexamers of the form GpApXpZpTpC, where the central XpZ step can be CpG, TpA, GpC, or ApT, has been studied by molecular modeling and molecular mechanics techniques. Interaction energies have also been calculated for the complexation of echinomycin with sequences containing the preferred central CpG step and different flanking base pairs. Besides, two more sets of sequences incorporating either 2,6-diaminopurine (DAP) or hypoxanthine in place of adenine or guanine, respectively, have been examined. The aim of this work was to evaluate the relative importance of hydrogen-bonding and stacking interactions in the association of echinomycin with DNA and further rationalize the experimental evidence. The results of these calculations are in consonance with available data from footprinting experiments and appear to support our previous hypothesis that, in addition to the crucial intermolecular hydrogen bonds in the central region, the stacking interactions involving the quinoxaline-2-carboxamide chromophores of the drug and the DNA base pairs play an important role in modulating the binding specificity of this bisintercalating antitumor antibiotic. This is most clearly seen when sequences with similar minor-groove environments are compared (e.g. CpI vs TpA or CpG vs TpDAP). The dipole moment of N-methylquinoxaline-2-carboxamide has been measured (mu = 4.15 +/- 0.03 D) and compares very well with the calculated value (mu = 4.14 D). The fact that G:C, I:C, A:T, and DAP:T base pairs are shown to be endowed with distinct van der Waals and electrostatic stacking properties with respect to this heteroaromatic ring system could have important implications for the design of novel DNA mono- and bis-intercalating agents.
PMID: 8201593, UI: 94260518
[Supporting Information]
Rational modification of human synovial fluid phospholipase A2 inhibitors.
Pisabarro MT, Ortiz AR, Palomer A, Cabre F, García L, Wade RC, Gago F, Mauleón D, Carganico G
European Molecular Biology Laboratory, Heidelberg, Germany / Departamento de Fisiología y Farmacología, Universidad de Alcalá, Alcalá de Henares, Madrid, Spain.
PMID: 8308860, UI: 94141887
J Pharm Sci 1993 Aug;82(8):794-798
Synthesis and structural, biochemical, and pharmacological study of 3 β-acyloxy-3
α-methoxycarbonyltropane derivatives.
Gálvez E, Izquierdo ML, Burgos C, Arias MS, Sanz-Aparicio J, Fonseca I, Gago F, Baldominos G, López P, Prieto JC
Departamento de Química Orgánica, Campus Universitario de Alcalá, Spain.
A series of 3 β-acyloxy-3 α-methoxycarbonyltropanes were synthesized and studied by 1H and 13C NMR spectroscopy, and the crystal structure of 3 α-methoxycarbonyl-3 β-pyridincarbonyloxytropane (5d) was determined by X-ray diffraction. In CDCl3 solution, compounds 5a-f display the same preferred conformation. The pyrrolidine and piperidine rings adopt an envelope conformation flattened at N-8 and a distorted chair conformation puckered at N-8 and flattened at C3, respectively, with the N-substituent in the equatorial position with respect to the piperidine ring. The pharmacological profile of one of these compounds makes it an adequate candidate for the design of novel GABAB antagonist agents.
PMID: 8377116, UI: 93389643
Molecular model of the interaction of bee venom phospholipase A2 with manoalide.
Ortiz AR, Pisabarro MT, Gago F
Departamento de Fisiología y Farmacología, Universidad de Alcalá de Henares, Madrid, Spain.
A molecular model of the interaction between manoalide (MLD) and bee venom phospholipase A2 (bv-PLA2) has been derived making use of a combination of computational methods. MLD was built in its open form and simulated by using molecular dynamics techniques. It is shown that the polar part of the molecule, which is thought to be the reactive region, is endowed with considerable conformational flexibility whereas the apolar region is rather rigid. The proposed active conformation of MLD and the main putative binding site for MLD on this enzyme were identified by matching potential energy GRID maps for both ligand and receptor with the chemical structure of the respective counterpart. The binding site is found in the C-terminal region of bv-PLA2, forming part of the proposed interfacial surface for binding to aggregated substrates, and comprises two distinct regions: (i) a hydrophobic cavity delimited by the C-terminal β-sheet and the antiparallel β-sheet, which interacts with the apolar zone of MLD, and (ii) a cationic site made up of residues Arg-58 and Lys-94, which interacts with the polar
zone. Molecular dynamics and molecular orbital calculations indicate that the most likely initial reaction between MLD and bv-PLA2 is formation of a Schiff base between Lys-94 and the aldehyde generated upon opening of MLD's γ-lactone ring, supporting recent model reaction studies. The inhibition seems to be a consequence of the occupation by MLD of a site overlapping a phosphocholine binding site in bv-PLA2 presumably involved in the interface desolvation process. The present model represents a starting point for further structural studies on the mechanism of phospholipases A2 inactivation by MLD and MLD-like compounds.
PMID: 8515424, UI: 93294800
A molecular dynamics study of the bis-intercalation complexes of echinomycin with d(ACGT)2 and d(TCGA)2: rationale for sequence-specific Hoogsteen base pairing.
Gallego J, Ortiz AR, Gago F
Departamento de Fisiología y Farmacología, Universidad de Alcalá de Henares, Madrid, Spain.
The behavior of the complexes of echinomycin with the DNA tetramers d(ACGT)2 and d(TCGA)2, in which the terminal AT base pairs are in either a Hoogsteen or a Watson-Crick conformation, has been explored by molecular dynamics taking into account experimental data from NMR studies (Gao and Patel. Biochemistry 1988, 27, 1744-1751). The DNA binding specificity of echinomycin appears to be the result of a subtle balance between stabilizing and destabilizing forces. Among the former is a number of hydrogen bonds between the alanine residues of echinomycin and both the N3 and 2-amino groups of the guanine bases which decisively determine the strong affinity of the antibiotic for CpG steps. On the other hand, there appears to be an unfavorable dipolar interaction between the chromophores of the antibiotic and the CpG step. This electrostatic component of the stacking interactions also contributes to explaining the conformational preferences of the flanking sequences: upon Hoogsteen pairing, the dipole moment of an AT base pair is found to increase significantly and alter its relative orientation. In the d(ACGT)2:echinomycin complex, this arrangement helps to improve the stacking interactions with the quinoxaline-2-carboxamide system, but would lead to unfavorable dipolar interactions in the d(TCGA)2 complex. The bearing of these findings on the binding of echinomycin to several
sequences as well as on the altered binding selectivity of other members of the quinoxaline family of antibiotics is also discussed.
PMID: 8496924, UI: 93267601
Three-dimensional structure of omega-conotoxin GVIA determined by 1H NMR.
Sevilla P, Bruix M, Santoro J, Gago F, García AG, Rico M
Dpto. Química Física II, Facultad de Farmacia, Universidad Complutense, Madrid, Spain / Departamento de Fisiología y Farmacología, Universidad de Alcalá, Alcalá de Henares, Madrid, Spain.
omega-Conotoxin GVIA, a peptide of 27 amino acid residues and three disulfide bridges, has been studied by NMR techniques. The complete assignment
of the corresponding proton NMR spectra was performed by two-dimensional sequence specific methods at 288 K and pH 3.5. On the basis of 169 distance
restraints derived from this analysis, the three-dimensional structure was obtained. A total of 30 initial structures were generated by distance
geometry methods and further refined by restrained energy minimization techniques yielding a final set of 8 structures. The mean root-mean-square
deviation between each of the 8 structures and the mean atomic coordinates for all residues is 0.82 +/- 0.06 Å for the backbone atoms and 1.45 +/- 0.18 Å for all non-H atoms. The structure shows a globular folding pattern that is stabilized by the three disulfide linkages and a number of intramolecular hydrogen bonds. A total of 14 hydroxyl groups are found at the periphery fully exposed to the solvent. These groups, together with the charged side chains of Lys and Arg residues emerging radially from the peptide core, provide specific recognition elements for the interaction of this toxin with neuronal calcium channels.
PMID: 8343203, UI: 93282829
Implications of a consensus recognition site for phosphatidylcholine separate from the active site in cobra venom phospholipases A2.
Ortiz AR, Pisabarro MT, Gallego J, Gago F
Departamento de Fisiología y Farmacología, Universidad de Alcalá de Henares, Madrid, Spain.
A model structure of Naja naja kaouthia cobra venom phospholipase A2 has been constructed by utilizing molecular modeling techniques. Analysis of the model and available biochemical data reveal the presence in this enzyme of a putative recognition site for choline derivatives in loop 57-70 made up of residues Trp-61, Tyr-63, Phe-64, and Lys-65, together with Glu-55. The magnitude and shape of the electrostatic potential in this binding site are approximately 80% similar to that in the McPC603 antibody binding site specifically recognizing phosphocholine. Docking studies indicate that the recognition site we now describe and the phosphocholine head of an n-alkylphosphocholine molecule are complementary both sterically and electronically, mainly due to anion-cation and cation-pi interactions. Moreover, binding enthalpies of n-heptylphosphocholine to this site are found to parallel the catalytic rate of pancreatic, mutant pancreatic, and cobra venom phospholipase A2 enzymes acting on dihexanoylphosphatidylcholine micelles, suggesting that it behaves as an activator site. This proposal is in keeping with the "dual phospholipid" model put forward to account for the phenomenon of interfacial activation. This novel site is also shown to be able to discriminate choline derivatives from ethanolamine derivatives, in accord with experimental data. On the basis of the results obtained, two functions are assigned to this putative activator site: (i) desolvation of the lipid-enzyme interface, particularly the surroundings of tyrosine
at position 69 (Tyr-63), and (ii) opening of the entrance to the active site by means of a conformational change of Tyr-63 whose chi 2 angle rotates
nearly 60 degrees.
PMID: 1550815, UI: 92198902
Anticancer Drug Des 1991 Feb;6(1):59-70
A prototype bioreductive DNA groove binding ligand.
Haworth IS, Burt C, Gago F, Reynolds CA, Richards WG
Physical Chemistry Laboratory, Oxford, UK.
Molecular mechanics calculations have been used to evaluate the potential bioreductive behaviour of several DNA minor groove binding ligands containing quinone/hydroquinone redox systems. The proposed structures are analogues of the Hoechst 33258 molecule with modifications of the benzimidazole rings. Binding energies of simple analogues indicate the reduced forms bind more strongly to the DNA minor groove. N-methylation of the imidazole ring(s) produces structures which can form extended quinone methides. These also show stronger binding in the reduced form and it is speculated that such structures might provide a basis for the design of groove binding ligands which will act as bioreductive alkylating agents.
PMID: 1707627, UI: 91197346
Netropsin binding to poly[d(IC)]·poly[d(IC)] and poly[d(GC)]·poly[d(GC)]: a computer simulation.
Gago F, Richards WG
Oxford Centre for Molecular Sciences, Oxford University, UK.
The thermodynamic cycle perturbation approach has been used to calculate the difference in the free energy of binding of netropsin to two different DNA molecules. In the computer simulations, all the inosine residues have been gradually 'mutated' into guanosine in a DNA dodecamer and in a complex of the same dodecamer with netropsin. The difference in binding free energy of about 4.3 kcal mol-1 agrees well with the experimentally determined value of 4.0 kcal mol-1. One structural determinant of the specificity seems to be the width of the minor groove in the two complexes.
PMID: 2156149, UI: 90190629
The binding of benzenesulfonamides to carbonic anhydrase enzyme. A molecular mechanics study and quantitative structure-activity relationships.
Menziani MC, De Benedetti PG, Gago F, Richards WG
Physical Chemistry Department, Oxford University, U.K.
Molecular mechanics methods have been applied to study the interaction between a series of 20 deprotonated benzenesulfonamides and the enzyme carbonic anhydrase. The different contributions to the binding energy have been evaluated and correlated with experimental inhibition data and molecular orbital indices of the sulfonamides in their bound conformation. The results suggest that the discrimination shown by the enzyme toward these inhibitors is dominated by the short-range van der Waals forces.
PMID: 2709382, UI: 89216826
The binding of nonintercalative drugs to alternating DNA sequences.
Gago F, Reynolds CA, Richards WG
Physical Chemistry Department, Oxford University, United Kingdom.
Molecular mechanics methods have been applied to suggest possible models for netropsin and related compounds binding to two different sequences of DNA, namely poly[d(AT)].poly[d(AT)] and poly[d(GC)].poly[d(GC)], and to evaluate the different contributions to the binding affinities of these compounds in the ethidium displacement assay. The geometries found after energy refinement suggest that one of the reasons for the selectivity of binding of these agents to A + T-rich DNA regions could be the different widths of the minor groove of the double strand of DNA found in the complexes of these drugs with both DNA sequences.
PMID: 2465487, UI: 89143479
One left-handed strand in DNA-oligonucleotide complexes?
Gago F, Richards WG
Physical Chemistry Laboratory, Oxford, England.
A single strand of oligonucleotide can bind to double helical DNA under certain conditions. This must involve some unwinding of the original double helix in a process leading to the formation of a three-stranded region. The free energy for such an entropically unlikely reaction may come from a change in the degree of supercoiling of the original DNA. The conformation of the triple strand is investigated here using computer graphics and molecular mechanics calculations. It is suggested that on binding the oligonucleotide (strand 3) to two paired strands (1 and 2) in a supercoiled DNA molecule, strand 2 might adopt a left-handed conformation whilst strand 1 and strand 3 pair in the normal Watson-Crick B-configuration.
PMID: 2914609, UI: 89121083
J Chromatogr 1988 Sep 30;449(1):95-101
Group contributions to hydrophobicity and elution behaviour of pyridine derivatives in reversed-phase high-performance liquid chromatography.
Gago F, Alvarez-Builla J, Elguero J
Department of Pharmacology, University of Alcalá de Henares, Madrid, Spain.
The mean hydrophobic contributions, π*m, of substituents in a sample of 34 monosubstituted pyridines has been determined by using reversed-phase high-performance liquid chromatography. The values are compared with those obtained for benzene.
PMID: 3235585, UI: 89175262


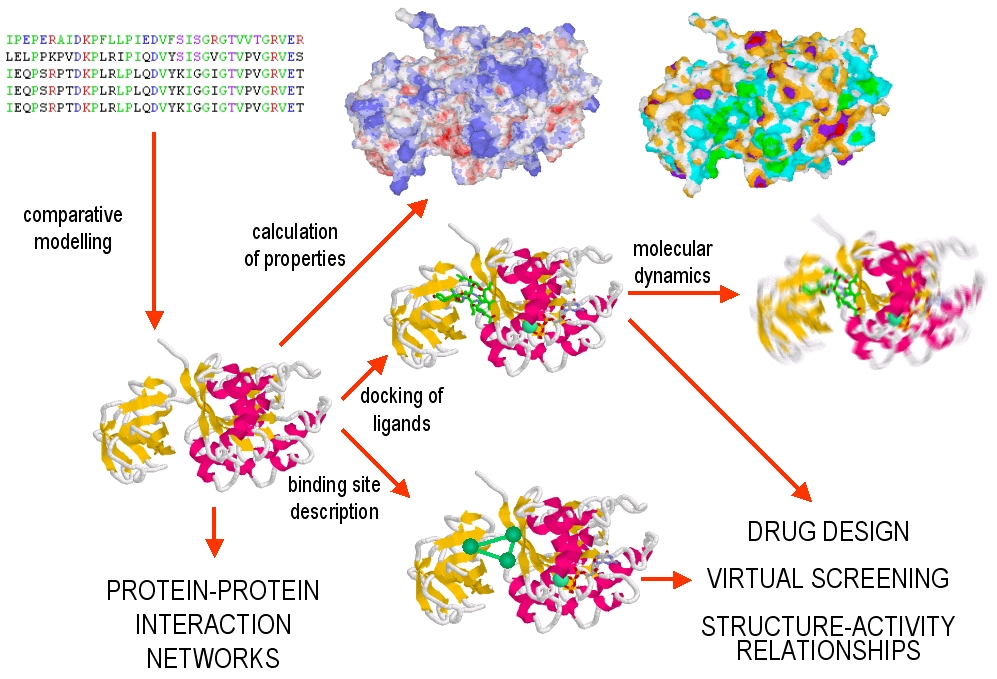
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}